Using pycisTopic on human cerebellum single-cell multiome data
[1]:
import os
os.chdir("/staging/leuven/stg_00002/lcb/sdewin/PhD/python_modules/pycisTopic_polars_tutorial")
[2]:
import pycisTopic
pycisTopic.__version__
[2]:
'2.0a0'
Downloading data
The data used for this tutorial is freely accessibile and can be downloaded from here, processed loom files can be found on Scope data can be explored on UCSC genome browser as well.
[5]:
!mkdir -p data
!wget -O data/fragments.tsv.gz https://cf.10xgenomics.com/samples/cell-arc/1.0.0/human_brain_3k/human_brain_3k_atac_fragments.tsv.gz
!wget -O data/fragments.tsv.gz.tbi https://cf.10xgenomics.com/samples/cell-arc/1.0.0/human_brain_3k/human_brain_3k_atac_fragments.tsv.gz.tbi
!wget -O data/cell_data.tsv https://raw.githubusercontent.com/aertslab/pycisTopic/polars/data/cell_data_human_cerebellum.tsv
--2024-02-29 17:02:20-- https://cf.10xgenomics.com/samples/cell-arc/1.0.0/human_brain_3k/human_brain_3k_atac_fragments.tsv.gz
Resolving cf.10xgenomics.com (cf.10xgenomics.com)... 2606:4700::6812:ad, 2606:4700::6812:1ad, 104.18.1.173, ...
Connecting to cf.10xgenomics.com (cf.10xgenomics.com)|2606:4700::6812:ad|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 1647677988 (1.5G) [text/tab-separated-values]
Saving to: ‘data/fragments.tsv.gz’
data/fragments.tsv. 100%[===================>] 1.53G 134MB/s in 9.6s
2024-02-29 17:02:30 (164 MB/s) - ‘data/fragments.tsv.gz’ saved [1647677988/1647677988]
--2024-02-29 17:02:30-- https://cf.10xgenomics.com/samples/cell-arc/1.0.0/human_brain_3k/human_brain_3k_atac_fragments.tsv.gz.tbi
Resolving cf.10xgenomics.com (cf.10xgenomics.com)... 2606:4700::6812:1ad, 2606:4700::6812:ad, 104.18.1.173, ...
Connecting to cf.10xgenomics.com (cf.10xgenomics.com)|2606:4700::6812:1ad|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 944619 (922K) [binary/octet-stream]
Saving to: ‘data/fragments.tsv.gz.tbi’
data/fragments.tsv. 100%[===================>] 922.48K --.-KB/s in 0.02s
2024-02-29 17:02:30 (37.7 MB/s) - ‘data/fragments.tsv.gz.tbi’ saved [944619/944619]
--2024-02-29 17:02:30-- https://raw.githubusercontent.com/aertslab/pycisTopic/polars/data/cell_data_human_cerebellum.tsv
Resolving raw.githubusercontent.com (raw.githubusercontent.com)... 2606:50c0:8001::154, 2606:50c0:8000::154, 2606:50c0:8002::154, ...
Connecting to raw.githubusercontent.com (raw.githubusercontent.com)|2606:50c0:8001::154|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 309204 (302K) [text/plain]
Saving to: ‘data/cell_data.tsv’
data/cell_data.tsv 100%[===================>] 301.96K --.-KB/s in 0.02s
2024-02-29 17:02:31 (17.6 MB/s) - ‘data/cell_data.tsv’ saved [309204/309204]
Set up
Let’s make some directories to store the output of pycisTopic.
[3]:
import os
out_dir = "outs"
os.makedirs(out_dir, exist_ok = True)
Define a dictionary mapping sample ids ("10x_multiome_brain") to fragment files ("data/fragments.tsv.gz").
Multiple entries can be added to this dictionary in case you have multiple samples.
Note:
pycisTopic will automatically append sample ids to barcodes to avoid barcode collisions between samples!
[4]:
fragments_dict = {
"10x_multiome_brain": "data/fragments.tsv.gz"
}
Getting pseudobulk profiles from cell annotations
In this tutorial we assume we are analyzing the scATAC-seq data from a multiome dataset, which allows to easily get the cell annotations from the scRNA-seq analysis. In case of independent scATAC-seq data, the cell annotation can also be obtained from alternative methods, such as unnanotated/preliminary clustering analysis (using predefined regions, for example SCREEN for mouse and human). In the later case, you can skip this section and use bulk regions as input to the QC step.
First we read the barcode-to-cell type annotation as a pd.DataFrame.
The index of the dataframe should correspond to the barcode column in the fragments.tsv.gz file (the sample-id can be optionally appended to the barcode using a split_pattern in this case "-") and must contain a column containing the sample id. Those sample ids should match with the sample ids defined in the fragments dictionary defined above.
Alternatively, a column name "barcode" can be added the metadata containing cell barcodes (without any suffixes!). In this case the index of the dataframe will not be used.
[5]:
import pandas as pd
cell_data = pd.read_table("data/cell_data.tsv", index_col = 0)
cell_data.head()
[5]:
| VSN_cell_type | VSN_leiden_res0.3 | VSN_leiden_res0.6 | VSN_leiden_res0.9 | VSN_leiden_res1.2 | VSN_sample_id | Seurat_leiden_res0.6 | Seurat_leiden_res1.2 | Seurat_cell_type | |
|---|---|---|---|---|---|---|---|---|---|
| AAACAGCCATTATGCG-1-10x_multiome_brain | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_1 (1) | MOL_B_3 (6) | 10x_multiome_brain | NFOL (1) | MOL (1) | MOL |
| AAACCAACATAGACCC-1-10x_multiome_brain | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_3 (5) | MOL_B_4 (4) | 10x_multiome_brain | NFOL (1) | NFOL (3) | NFOL |
| AAACCGAAGATGCCTG-1-10x_multiome_brain | INH_VIP | INH_VIP (6) | INH_VIP (8) | INH_VIP (8) | INH_VIP (10) | 10x_multiome_brain | INH_VIP (7) | INH_VIP (6) | INH_VIP |
| AAACCGAAGTTAGCTA-1-10x_multiome_brain | MOL_A | MOL_A (1) | MOL_A_2 (1) | MOL_A_1 (0) | MOL_A_2 (0) | 10x_multiome_brain | NFOL (1) | NFOL (3) | NFOL |
| AAACCGCGTTAGCCAA-1-10x_multiome_brain | MGL | MGL (7) | MGL (10) | MGL (10) | MGL (12) | 10x_multiome_brain | MGL (8) | MGL (9) | MGL |
We will also need the size of each chromosome, this can be downloaded from the UCSC databases.
[6]:
chromsizes = pd.read_table(
"http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.chrom.sizes",
header = None,
names = ["Chromosome", "End"]
)
chromsizes.insert(1, "Start", 0)
chromsizes.head()
[6]:
| Chromosome | Start | End | |
|---|---|---|---|
| 0 | chr1 | 0 | 248956422 |
| 1 | chr2 | 0 | 242193529 |
| 2 | chr3 | 0 | 198295559 |
| 3 | chr4 | 0 | 190214555 |
| 4 | chr5 | 0 | 181538259 |
Now we a ready to generate pseudobulk ATAC-seq profiles.
This function will generate a fragments.tsv.gz and a bigwig file for each cell type defined by the variable parameter.
For each cell type:
The
fragments.tsv.gzcontains all fragments for barcodes of that cell type.A
bigwigfile will be generated for eachfragments.tsv.gzfile, this you can visualize in any genome browser.
[8]:
from pycisTopic.pseudobulk_peak_calling import export_pseudobulk
os.makedirs(os.path.join(out_dir, "consensus_peak_calling"), exist_ok = True)
os.makedirs(os.path.join(out_dir, "consensus_peak_calling/pseudobulk_bed_files"), exist_ok = True)
os.makedirs(os.path.join(out_dir, "consensus_peak_calling/pseudobulk_bw_files"), exist_ok = True)
bw_paths, bed_paths = export_pseudobulk(
input_data = cell_data,
variable = "VSN_cell_type",
sample_id_col = "VSN_sample_id",
chromsizes = chromsizes,
bed_path = os.path.join(out_dir, "consensus_peak_calling/pseudobulk_bed_files"),
bigwig_path = os.path.join(out_dir, "consensus_peak_calling/pseudobulk_bw_files"),
path_to_fragments = fragments_dict,
n_cpu = 10,
normalize_bigwig = True,
temp_dir = "/tmp",
split_pattern = "-"
)
2024-03-05 13:59:29,358 cisTopic INFO Splitting fragments by cell type.
2024-03-05 14:00:46,456 cisTopic INFO generating bigwig files
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/joblib/externals/loky/process_executor.py:752: UserWarning: A worker stopped while some jobs were given to the executor. This can be caused by a too short worker timeout or by a memory leak.
warnings.warn(
We will need the paths to the bed files later on, so let’s save them to disk.
[9]:
with open(os.path.join(out_dir, "consensus_peak_calling/bw_paths.tsv"), "wt") as f:
for v in bw_paths:
_ = f.write(f"{v}\t{bw_paths[v]}\n")
[10]:
with open(os.path.join(out_dir, "consensus_peak_calling/bed_paths.tsv"), "wt") as f:
for v in bed_paths:
_ = f.write(f"{v}\t{bed_paths[v]}\n")
Inferring consensus peaks
Next we will use MACS to call peaks for each pseudobulk fragments.tsv.gz file.
[11]:
bw_paths = {}
with open(os.path.join(out_dir, "consensus_peak_calling/bw_paths.tsv")) as f:
for line in f:
v, p = line.strip().split("\t")
bw_paths.update({v: p})
[12]:
bed_paths = {}
with open(os.path.join(out_dir, "consensus_peak_calling/bed_paths.tsv")) as f:
for line in f:
v, p = line.strip().split("\t")
bed_paths.update({v: p})
[14]:
from pycisTopic.pseudobulk_peak_calling import peak_calling
macs_path = "macs2"
os.makedirs(os.path.join(out_dir, "consensus_peak_calling/MACS"), exist_ok = True)
narrow_peak_dict = peak_calling(
macs_path = macs_path,
bed_paths = bed_paths,
outdir = os.path.join(os.path.join(out_dir, "consensus_peak_calling/MACS")),
genome_size = 'hs',
n_cpu = 10,
input_format = 'BEDPE',
shift = 73,
ext_size = 146,
keep_dup = 'all',
q_value = 0.05,
_temp_dir = '/scratch/leuven/330/vsc33053/ray_spill'
)
2024-02-29 18:05:49,348 INFO worker.py:1724 -- Started a local Ray instance.
(macs_call_peak_ray pid=1232492) 2024-02-29 18:05:51,892 cisTopic INFO Calling peaks for AST_CER with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/AST_CER.fragments.tsv.gz --name AST_CER --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232485) 2024-02-29 18:05:51,919 cisTopic INFO Calling peaks for GC with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/GC.fragments.tsv.gz --name GC --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232488) 2024-02-29 18:05:51,975 cisTopic INFO Calling peaks for OPC with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/OPC.fragments.tsv.gz --name OPC --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232483) 2024-02-29 18:05:51,936 cisTopic INFO Calling peaks for ASTP with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/ASTP.fragments.tsv.gz --name ASTP --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232484) 2024-02-29 18:05:51,888 cisTopic INFO Calling peaks for INH_PVALB with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/INH_PVALB.fragments.tsv.gz --name INH_PVALB --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232489) 2024-02-29 18:05:51,895 cisTopic INFO Calling peaks for MGL with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/MGL.fragments.tsv.gz --name MGL --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232490) 2024-02-29 18:05:51,975 cisTopic INFO Calling peaks for INH_VIP with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/INH_VIP.fragments.tsv.gz --name INH_VIP --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232491) 2024-02-29 18:05:51,896 cisTopic INFO Calling peaks for MOL_A with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/MOL_A.fragments.tsv.gz --name MOL_A --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232486) 2024-02-29 18:05:51,961 cisTopic INFO Calling peaks for PURK with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/PURK.fragments.tsv.gz --name PURK --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232487) 2024-02-29 18:05:51,915 cisTopic INFO Calling peaks for MOL_B with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/MOL_B.fragments.tsv.gz --name MOL_B --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232484) 2024-02-29 18:05:58,586 cisTopic INFO INH_PVALB done!
(macs_call_peak_ray pid=1232484) 2024-02-29 18:05:58,594 cisTopic INFO Calling peaks for INH_SST with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/INH_SST.fragments.tsv.gz --name INH_SST --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232486) 2024-02-29 18:06:12,711 cisTopic INFO PURK done!
(macs_call_peak_ray pid=1232486) 2024-02-29 18:06:12,725 cisTopic INFO Calling peaks for AST with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/AST.fragments.tsv.gz --name AST --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232483) 2024-02-29 18:06:21,180 cisTopic INFO ASTP done!
(macs_call_peak_ray pid=1232483) 2024-02-29 18:06:21,201 cisTopic INFO Calling peaks for GP with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/GP.fragments.tsv.gz --name GP --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232489) 2024-02-29 18:06:21,581 cisTopic INFO MGL done!
(macs_call_peak_ray pid=1232489) 2024-02-29 18:06:21,604 cisTopic INFO Calling peaks for ENDO with macs2 callpeak --treatment outs/consensus_peak_calling/pseudobulk_bed_files/ENDO.fragments.tsv.gz --name ENDO --outdir outs/consensus_peak_calling/MACS --format BEDPE --gsize hs --qvalue 0.05 --nomodel --shift 73 --extsize 146 --keep-dup all --call-summits --nolambda
(macs_call_peak_ray pid=1232489) 2024-02-29 18:06:28,838 cisTopic INFO ENDO done!
(macs_call_peak_ray pid=1232486) 2024-02-29 18:06:34,004 cisTopic INFO AST done!
(macs_call_peak_ray pid=1232490) 2024-02-29 18:06:46,068 cisTopic INFO INH_VIP done!
(macs_call_peak_ray pid=1232488) 2024-02-29 18:06:47,921 cisTopic INFO OPC done!
(macs_call_peak_ray pid=1232484) 2024-02-29 18:06:52,269 cisTopic INFO INH_SST done!
(macs_call_peak_ray pid=1232491) 2024-02-29 18:07:00,088 cisTopic INFO MOL_A done!
(macs_call_peak_ray pid=1232483) 2024-02-29 18:07:04,270 cisTopic INFO GP done!
(macs_call_peak_ray pid=1232487) 2024-02-29 18:07:05,433 cisTopic INFO MOL_B done!
(macs_call_peak_ray pid=1232485) 2024-02-29 18:07:15,367 cisTopic INFO GC done!
(macs_call_peak_ray pid=1232492) 2024-02-29 18:07:17,439 cisTopic INFO AST_CER done!
Finally, it is time to derive the consensus peaks. To do so, we use the TGCA iterative peak filtering approach. First, each summit is extended a peak_half_width in each direction and then we iteratively filter out less significant peaks that overlap with a more significant one. During this procedure peaks will be merged and depending on the number of peaks included into them, different processes will happen:
1 peak: The original peak region will be kept
2 peaks: The original peak region with the highest score will be kept
3 or more peaks: The orignal peak region with the most significant score will be taken, and all the original peak regions in this merged peak region that overlap with the significant peak region will be removed. The process is repeated with the next most significant peak (if it was not removed already) until all peaks are processed.
This proccess will happen twice, first for each pseudobulk peaks; and after peak score normalization, to process all peaks together.
[16]:
from pycisTopic.iterative_peak_calling import get_consensus_peaks
# Other param
peak_half_width=250
path_to_blacklist="pycisTopic/blacklist/hg38-blacklist.v2.bed"
# Get consensus peaks
consensus_peaks = get_consensus_peaks(
narrow_peaks_dict = narrow_peak_dict,
peak_half_width = peak_half_width,
chromsizes = chromsizes,
path_to_blacklist = path_to_blacklist)
2024-02-29 18:07:54,059 cisTopic INFO Extending and merging peaks per class
2024-02-29 18:08:50,783 cisTopic INFO Normalizing peak scores
2024-02-29 18:08:51,331 cisTopic INFO Merging peaks
2024-02-29 18:09:59,689 cisTopic INFO Done!
[17]:
consensus_peaks.to_bed(
path = os.path.join(out_dir, "consensus_peak_calling/consensus_regions.bed"),
keep =True,
compression = 'infer',
chain = False)
QC
The next step is to perform QC for the scATAC-seq samples (in this case, only one run). There are several measurements and visualizations performed in this step:
Barcode rank plot
Duplication rate
Insertion size
TSS enrichment
Fraction of Reads In Peaks (FRIP)
To calculate the TSS enrichment we need to provide TSS annotations. You can easily download them via the pycistopic tss get_tss command.
In case you are unsure which column name is used by Ensembl to specify gene names in their databases, run the pycistopic tss gene_annotation_list and grep for your species.
[14]:
!pycistopic tss gene_annotation_list | grep Human
hsapiens_gene_ensembl Human genes (GRCh38.p14)
[17]:
!mkdir -p outs/qc
!pycistopic tss get_tss \
--output outs/qc/tss.bed \
--name "hsapiens_gene_ensembl" \
--to-chrom-source ucsc \
--ucsc hg38
- Get TSS annotation from Ensembl BioMart with the following settings:
- biomart_name: "hsapiens_gene_ensembl"
- biomart_host: "http://www.ensembl.org"
- transcript_type: ['protein_coding']
- use_cache: True
- Getting chromosome sizes and alias mapping for "hg38" from UCSC.
- Update chromosome names in TSS annotation to "ucsc" chromosome names.
- Writing TSS annotation BED file to "outs/qc/tss.bed".
[19]:
!head outs/qc/tss.bed | column -t
# Chromosome Start End Gene Score Strand Transcript_type
chrM 3306 3307 MT-ND1 . + protein_coding
chrM 4469 4470 MT-ND2 . + protein_coding
chrM 5903 5904 MT-CO1 . + protein_coding
chrM 7585 7586 MT-CO2 . + protein_coding
chrM 8365 8366 MT-ATP8 . + protein_coding
chrM 8526 8527 MT-ATP6 . + protein_coding
chrM 9206 9207 MT-CO3 . + protein_coding
chrM 10058 10059 MT-ND3 . + protein_coding
chrM 10469 10470 MT-ND4L . + protein_coding
Next, let’s calculate the QC metrics using the pycistopic qc command.
[24]:
!pycistopic qc \
--fragments data/fragments.tsv.gz \
--regions outs/consensus_peak_calling/consensus_regions.bed \
--tss outs/qc/tss.bed \
--output outs/qc/10x_multiome_brain
In case you have multiple samples, you can run the QC step in parallel as follows.
[ ]:
regions_bed_filename = os.path.join(out_dir, "consensus_peak_calling/consensus_regions.bed")
tss_bed_filename = os.path.join(out_dir, "qc", "tss.bed")
pycistopic_qc_commands_filename = "pycistopic_qc_commands.txt"
# Create text file with all pycistopic qc command lines.
with open(pycistopic_qc_commands_filename, "w") as fh:
for sample, fragment_filename in fragments_dict.items():
print(
"pycistopic qc",
f"--fragments {fragment_filename}",
f"--regions {regions_bed_filename}",
f"--tss {tss_bed_filename}",
f"--output {os.path.join(out_dir, "qc")}/{sample}",
sep=" ",
file=fh,
)
And run the following in a command line environment.
cat pycistopic_qc_commands.txt | parallel -j 4 {}
Finally, we can visualize sample level statistics.
These include:
Barcode rank plot: The barcode rank plot shows the distribution of non-duplicate reads and which barcodes were inferred to be associated with cells. A steep drop-off (‘knee’) is indicative of good separation between the cell-associated barcodes and the barcodes associated with empty partitions.
Insertion size: ATAC-seq requires a proper pair of Tn5 transposase cutting events at the ends of DNA. In the nucleosome-free open chromatin regions, many molecules of Tn5 can kick in and chop the DNA into small pieces; around nucleosome-occupied regions, and Tn5 can only access the linker regions. Therefore, in a good ATAC-seq library, you should expect to see a sharp peak at the <100 bp region (open chromatin), and a peak at ~200bp region (mono-nucleosome), and other larger peaks (multi-nucleosomes). A clear nucleosome pattern indicates a good quality of the experiment.
Sample TSS enrichment: The TSS enrichment calculation is a signal to noise calculation. The reads around a reference set of TSSs are collected to form an aggregate distribution of reads centered on the TSSs and extending to 1000 bp in either direction (for a total of 2000bp). This distribution is then normalized by taking the average read depth in the 100 bps at each of the end flanks of the distribution (for a total of 200bp of averaged data) and calculating a fold change at each position over that average read depth. This means that the flanks should start at 1, and if there is high read signal at transcription start sites (highly open regions of the genome) there should be an increase in signal up to a peak in the middle.
[6]:
from pycisTopic.plotting.qc_plot import plot_sample_stats, plot_barcode_stats
import matplotlib.pyplot as plt
[7]:
for sample_id in fragments_dict:
fig = plot_sample_stats(
sample_id = sample_id,
pycistopic_qc_output_dir = "outs/qc"
)
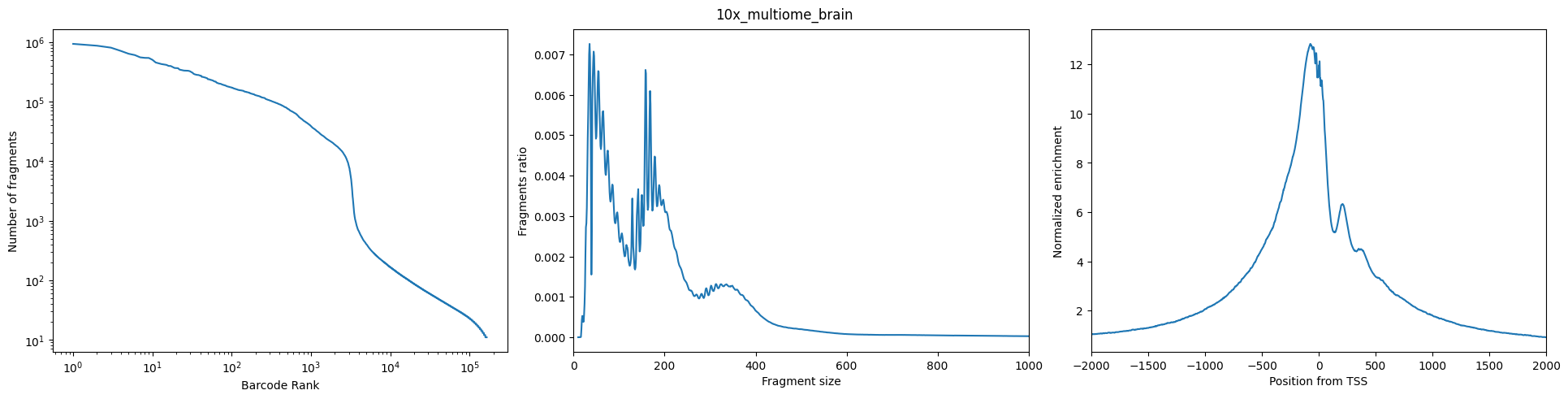
We can also visualize barcode level statistics.
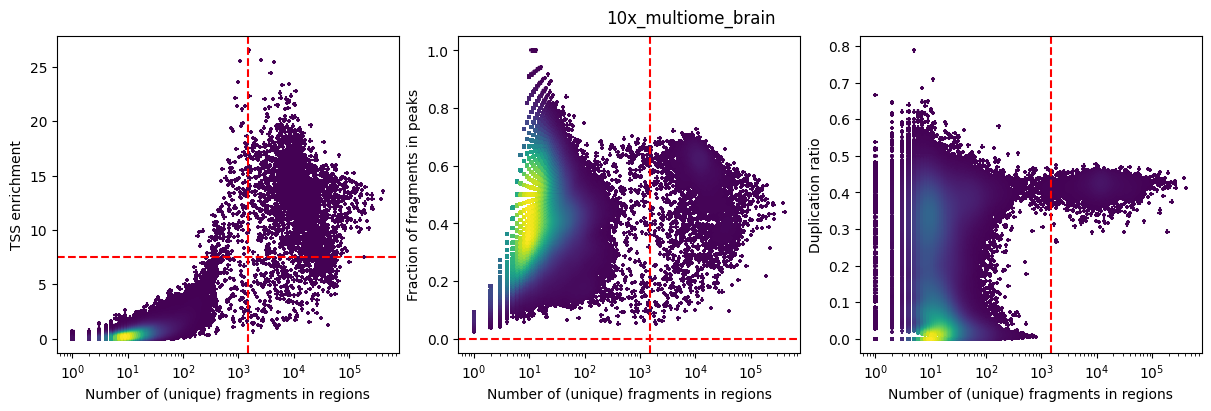
These statistics can be used to filter cell barcodes to retain only high quality cells.
Note:
The pycistopic qc command will determine automatic thresholds for the minimum number of unique number of fragments and the minumum TSS enrichment. In case you want to change these thresholds or want to threhold based on FRIP, you can provide manually defined thresholds using the parameters: - unique_fragments_threshold - tss_enrichment_threshold - frip_threshold
In this case we will use the automatically defined thresholds, please manualy inspect the quality metrics to make sure these thresholds are valid!
The barcode level statistics include:
Total number of (unique) fragments
TSS enrichment: The score at position in the TSS enrichmen score for for each barcode (at position 0, the TSS). Noisy cells will have a low TSS enrichment.
FRIP: The fraction of reads in peaks for each barcode. Noisy cells have low FRIP values. However, this filter should be used with nuance, as it depends on the quality of the original peaks. For example, if there is a rare population in the sample, its specific peaks may be missed by peak calling algorithms, causing a decrease in their FRIP values.
[6]:
from pycisTopic.qc import get_barcodes_passing_qc_for_sample
sample_id_to_barcodes_passing_filters = {}
sample_id_to_thresholds = {}
for sample_id in fragments_dict:
(
sample_id_to_barcodes_passing_filters[sample_id],
sample_id_to_thresholds[sample_id]
) = get_barcodes_passing_qc_for_sample(
sample_id = sample_id,
pycistopic_qc_output_dir = "outs/qc",
unique_fragments_threshold = None, # use automatic thresholding
tss_enrichment_threshold = None, # use automatic thresholding
frip_threshold = 0,
use_automatic_thresholds = True,
)
10x_multiome_brain:
Using automatic threshold for unique fragments: 1500.1880245466896
Using automatic threshold for TSS enrichment: 7.534788253503151
[9]:
for sample_id in fragments_dict:
fig = plot_barcode_stats(
sample_id = sample_id,
pycistopic_qc_output_dir = "outs/qc",
bc_passing_filters = sample_id_to_barcodes_passing_filters[sample_id],
detailed_title = False,
**sample_id_to_thresholds[sample_id]
)
Creating a cisTopic object
In this step we will create a cisTopic object. This involves generating a count matrix containing fragment counts over consensus peaks (see above) for each cell barcode passing the QC metrices defined above.
Blacklist regions will be removed from this count matrix.
[9]:
path_to_regions = os.path.join(out_dir, "consensus_peak_calling/consensus_regions.bed")
path_to_blacklist = "pycisTopic/blacklist/hg38-blacklist.v2.bed"
pycistopic_qc_output_dir = "outs/qc"
from pycisTopic.cistopic_class import create_cistopic_object_from_fragments
import polars as pl
cistopic_obj_list = []
for sample_id in fragments_dict:
sample_metrics = pl.read_parquet(
os.path.join(pycistopic_qc_output_dir, f'{sample_id}.fragments_stats_per_cb.parquet')
).to_pandas().set_index("CB").loc[ sample_id_to_barcodes_passing_filters[sample_id] ]
cistopic_obj = create_cistopic_object_from_fragments(
path_to_fragments = fragments_dict[sample_id],
path_to_regions = path_to_regions,
path_to_blacklist = path_to_blacklist,
metrics = sample_metrics,
valid_bc = sample_id_to_barcodes_passing_filters[sample_id],
n_cpu = 1,
project = sample_id,
split_pattern = '-'
)
cistopic_obj_list.append(cistopic_obj)
2024-03-06 10:15:48,250 cisTopic INFO Reading data for 10x_multiome_brain
2024-03-06 10:17:22,144 cisTopic INFO metrics provided!
2024-03-06 10:17:30,393 cisTopic INFO Counting fragments in regions
2024-03-06 10:18:32,026 cisTopic INFO Creating fragment matrix
2024-03-06 10:19:03,991 cisTopic INFO Converting fragment matrix to sparse matrix
2024-03-06 10:19:10,609 cisTopic INFO Removing blacklisted regions
2024-03-06 10:19:11,744 cisTopic INFO Creating CistopicObject
2024-03-06 10:19:13,305 cisTopic INFO Done!
In this case we only have one sample, so only one cisTopic object has been generated. If you would have multiple samples, you would need to run the merge() function on your cisTopic object list.
[10]:
cistopic_obj = cistopic_obj_list[0]
print(cistopic_obj)
CistopicObject from project 10x_multiome_brain with n_cells × n_regions = 2853 × 436234
[13]:
import pickle
pickle.dump(
cistopic_obj,
open(os.path.join(out_dir, "cistopic_obj.pkl"), "wb")
)
Adding metadata to a cisTopic object
We can add additional metadata (for regions or cells) to a cisTopic object. For example, let’s add the scRNA-seq data annotations. Missing values will be filled with Nan.
[14]:
import pandas as pd
cell_data = pd.read_table("data/cell_data.tsv", index_col = 0)
cell_data.head()
cistopic_obj.add_cell_data(cell_data, split_pattern='-')
pickle.dump(
cistopic_obj,
open(os.path.join(out_dir, "cistopic_obj.pkl"), "wb")
)
Warning: Some cells in this CistopicObject are not present in this cell_data. Values will be filled with Nan
[15]:
cistopic_obj.cell_data
[15]:
| cisTopic_nr_frag | cisTopic_log_nr_frag | cisTopic_nr_acc | cisTopic_log_nr_acc | sample_id | barcode_rank | total_fragments_count | log10_total_fragments_count | unique_fragments_count | log10_unique_fragments_count | ... | barcode | VSN_cell_type | VSN_leiden_res0.3 | VSN_leiden_res0.6 | VSN_leiden_res0.9 | VSN_leiden_res1.2 | VSN_sample_id | Seurat_leiden_res0.6 | Seurat_leiden_res1.2 | Seurat_cell_type | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CACCTCAGTTGTAAAC-1-10x_multiome_brain | 18300 | 4.262451 | 15555 | 4.19187 | 10x_multiome_brain | 1263 | 47941 | 4.680716 | 30052 | 4.477888 | ... | CACCTCAGTTGTAAAC-1 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| TGACTCCTCATCCACC-1-10x_multiome_brain | 100055 | 5.000239 | 62282 | 4.794363 | 10x_multiome_brain | 93 | 311527 | 5.493497 | 177932 | 5.250257 | ... | TGACTCCTCATCCACC-1 | AST_CER | AST_CER (2) | AST_CER (2) | AST_CER (2) | AST_CER_1 (7) | 10x_multiome_brain | BG (2) | BG (2) | BG |
| TTTCTCACATAAACCT-1-10x_multiome_brain | 32192 | 4.507748 | 27001 | 4.43138 | 10x_multiome_brain | 594 | 110849 | 5.044736 | 67109 | 4.826787 | ... | TTTCTCACATAAACCT-1 | GC | GC (4) | GC (5) | GC (6) | GC (8) | 10x_multiome_brain | GC (4) | GC (5) | GC |
| GTCCTCCCACACAATT-1-10x_multiome_brain | 88443 | 4.946663 | 58296 | 4.765639 | 10x_multiome_brain | 97 | 325923 | 5.513116 | 174752 | 5.242425 | ... | GTCCTCCCACACAATT-1 | AST_CER | AST_CER (2) | AST_CER (2) | AST_CER (2) | AST_CER_1 (7) | 10x_multiome_brain | BG (2) | BG (2) | BG |
| CTCCGTCCAGTTTGTG-1-10x_multiome_brain | 131110 | 5.117636 | 78509 | 4.894919 | 10x_multiome_brain | 54 | 405882 | 5.608401 | 232359 | 5.366161 | ... | CTCCGTCCAGTTTGTG-1 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| GCAGGTTGTCCAAATG-1-10x_multiome_brain | 2770 | 3.44248 | 2613 | 3.417139 | 10x_multiome_brain | 3258 | 7170 | 3.855580 | 4043 | 3.606811 | ... | GCAGGTTGTCCAAATG-1 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| AAGCTCCCAGCACCAT-1-10x_multiome_brain | 2180 | 3.338456 | 2040 | 3.30963 | 10x_multiome_brain | 3148 | 9905 | 3.995898 | 5832 | 3.765892 | ... | AAGCTCCCAGCACCAT-1 | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| CAGAATCTCCTCATGC-1-10x_multiome_brain | 1744 | 3.241546 | 1681 | 3.225568 | 10x_multiome_brain | 3340 | 4638 | 3.666424 | 2655 | 3.424228 | ... | CAGAATCTCCTCATGC-1 | MOL_B | MOL_B (0) | MOL_B_1 (0) | MOL_B_1 (1) | MOL_B_1 (1) | 10x_multiome_brain | MOL (0) | MOL (1) | MOL |
| TAGCCGGGTAACAGGG-1-10x_multiome_brain | 2674 | 3.427161 | 2414 | 3.382737 | 10x_multiome_brain | 3134 | 10310 | 4.013301 | 6033 | 3.780605 | ... | TAGCCGGGTAACAGGG-1 | ENDO | MOL_A (1) | MOL_A_2 (1) | MOL_A_1 (0) | ENDO (18) | 10x_multiome_brain | AST+ENDO (6) | ENDO (15) | ENDO |
| GTGCGCAGTGCTTAGA-1-10x_multiome_brain | 5859 | 3.767823 | 5515 | 3.741546 | 10x_multiome_brain | 2452 | 25196 | 4.401349 | 14736 | 4.168409 | ... | GTGCGCAGTGCTTAGA-1 | INH_SST | INH_SST (5) | INH_SST (7) | INH_SST (7) | INH_SST (9) | 10x_multiome_brain | INH_SST (5) | INH_SST (8) | INH_SST |
2853 rows × 31 columns
Running scrublet
Optionally, you can run also scrublet on the fragment count matrix to infer doublets from the scATAC-seq.
[17]:
import scrublet as scr
scrub = scr.Scrublet(cistopic_obj.fragment_matrix.T, expected_doublet_rate=0.1)
doublet_scores, predicted_doublets = scrub.scrub_doublets()
scrub.plot_histogram();
scrub.call_doublets(threshold=0.22)
scrub.plot_histogram();
scrublet = pd.DataFrame([scrub.doublet_scores_obs_, scrub.predicted_doublets_], columns=cistopic_obj.cell_names, index=['Doublet_scores_fragments', 'Predicted_doublets_fragments']).T
Preprocessing...
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/scrublet/helper_functions.py:252: RuntimeWarning: invalid value encountered in sqrt
CV_input = np.sqrt(b);
Simulating doublets...
Embedding transcriptomes using PCA...
Calculating doublet scores...
Automatically set threshold at doublet score = 0.66
Detected doublet rate = 0.2%
Estimated detectable doublet fraction = 1.1%
Overall doublet rate:
Expected = 10.0%
Estimated = 16.1%
Elapsed time: 26.8 seconds
Detected doublet rate = 22.4%
Estimated detectable doublet fraction = 66.6%
Overall doublet rate:
Expected = 10.0%
Estimated = 33.6%
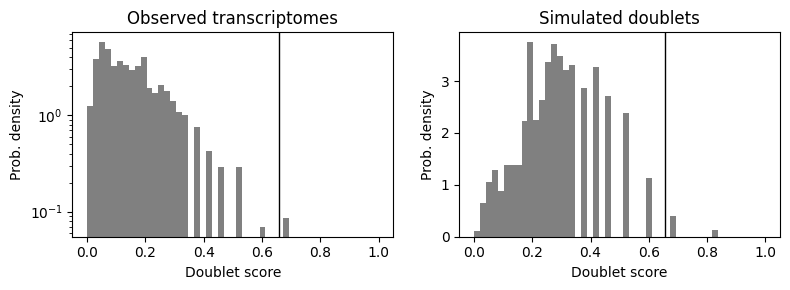
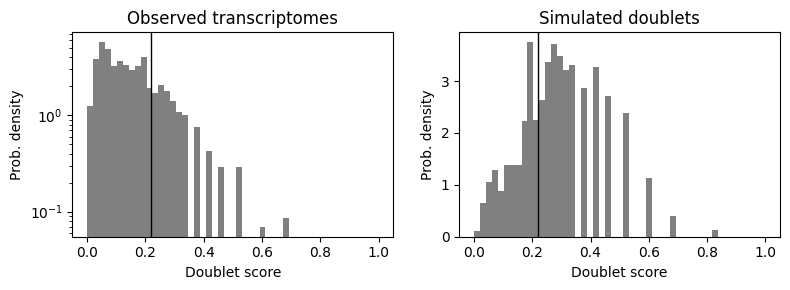
[18]:
cistopic_obj.add_cell_data(scrublet, split_pattern = '-')
sum(cistopic_obj.cell_data.Predicted_doublets_fragments == True)
[18]:
638
[19]:
pickle.dump(
cistopic_obj,
open(os.path.join(out_dir, "cistopic_obj.pkl"), "wb")
)
[20]:
# Remove doublets
singlets = cistopic_obj.cell_data[cistopic_obj.cell_data.Predicted_doublets_fragments == False].index.tolist()
# Subset cisTopic object
cistopic_obj_noDBL = cistopic_obj.subset(singlets, copy=True, split_pattern='-')
print(cistopic_obj_noDBL)
CistopicObject from project 10x_multiome_brain with n_cells × n_regions = 2215 × 436232
[21]:
pickle.dump(
cistopic_obj,
open(os.path.join(out_dir, "cistopic_obj.pkl"), "wb")
)
Run models
Next we will perform the actual topic modeling using LDA using a Collapsed Gibbs Sampler.
There are two functions that ca be used to perform the topic modeling, both producde similar results.
Serial LDA: The parallelization is done between models rather than within each model. Recommended for small-medium sized data sets in which several models with different number of topics are being tested. You can run these models with runCGSModels().
Parallel LDA with MALLET: The parallelization is done within each model. Recommended for large data sets where a few models with different number of topics are being tested. If working in a cluster, we recommed to submit a job per model so they can run simultaneously. You can run it with runCGSModelsMallet().
Here, we will use Mallet.
Note:
In order to be able to run Mallet you need the Mallet binary, these binaries can be downloaded from Github. You can also compile the binary from source, for more information please visit the Mallet Github repository.
[23]:
!wget https://github.com/mimno/Mallet/releases/download/v202108/Mallet-202108-bin.tar.gz
!tar -xf Mallet-202108-bin.tar.gz
--2024-03-06 10:35:09-- https://github.com/mimno/Mallet/releases/download/v202108/Mallet-202108-bin.tar.gz
Resolving github.com (github.com)... 140.82.121.4
Connecting to github.com (github.com)|140.82.121.4|:443... connected.
HTTP request sent, awaiting response... 302 Found
Location: https://objects.githubusercontent.com/github-production-release-asset-2e65be/18378040/6a3fdbe6-0d3f-4f99-add3-1f98129f43cb?X-Amz-Algorithm=AWS4-HMAC-SHA256&X-Amz-Credential=AKIAVCODYLSA53PQK4ZA%2F20240306%2Fus-east-1%2Fs3%2Faws4_request&X-Amz-Date=20240306T093319Z&X-Amz-Expires=300&X-Amz-Signature=441a3185ed2778aeb182bf8f470200f1c88411564e67493f8e82405ac057b350&X-Amz-SignedHeaders=host&actor_id=0&key_id=0&repo_id=18378040&response-content-disposition=attachment%3B%20filename%3DMallet-202108-bin.tar.gz&response-content-type=application%2Foctet-stream [following]
--2024-03-06 10:35:09-- https://objects.githubusercontent.com/github-production-release-asset-2e65be/18378040/6a3fdbe6-0d3f-4f99-add3-1f98129f43cb?X-Amz-Algorithm=AWS4-HMAC-SHA256&X-Amz-Credential=AKIAVCODYLSA53PQK4ZA%2F20240306%2Fus-east-1%2Fs3%2Faws4_request&X-Amz-Date=20240306T093319Z&X-Amz-Expires=300&X-Amz-Signature=441a3185ed2778aeb182bf8f470200f1c88411564e67493f8e82405ac057b350&X-Amz-SignedHeaders=host&actor_id=0&key_id=0&repo_id=18378040&response-content-disposition=attachment%3B%20filename%3DMallet-202108-bin.tar.gz&response-content-type=application%2Foctet-stream
Resolving objects.githubusercontent.com (objects.githubusercontent.com)... 185.199.111.133, 185.199.110.133, 185.199.109.133, ...
Connecting to objects.githubusercontent.com (objects.githubusercontent.com)|185.199.111.133|:443... connected.
HTTP request sent, awaiting response... 200 OK
Length: 7187546 (6.9M) [application/octet-stream]
Saving to: ‘Mallet-202108-bin.tar.gz’
Mallet-202108-bin.t 100%[===================>] 6.85M --.-KB/s in 0.05s
2024-03-06 10:35:09 (138 MB/s) - ‘Mallet-202108-bin.tar.gz’ saved [7187546/7187546]
[ ]:
!mkdir -p /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/
Because we don’t know yet what number of topics will be optimal for our dataset we will run several topic models, each with a different number of topics.
[27]:
os.environ['MALLET_MEMORY'] = '200G'
from pycisTopic.lda_models import run_cgs_models_mallet
# Configure path Mallet
mallet_path="Mallet-202108/bin/mallet"
# Run models
models=run_cgs_models_mallet(
cistopic_obj,
n_topics=[2, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50],
n_cpu=12,
n_iter=500,
random_state=555,
alpha=50,
alpha_by_topic=True,
eta=0.1,
eta_by_topic=False,
tmp_path="/scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial",
save_path="/scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial",
mallet_path=mallet_path,
)
2024-03-06 10:52:57,071 cisTopic INFO Formatting input to corpus
2024-03-06 10:52:57,690 gensim.corpora.dictionary INFO adding document #0 to Dictionary<0 unique tokens: []>
2024-03-06 10:53:23,393 gensim.corpora.dictionary INFO built Dictionary<436234 unique tokens: ['0', '1', '2', '3', '4']...> from 2853 documents (total 46261622 corpus positions)
2024-03-06 10:53:23,395 cisTopic INFO Running model with 2 topics
2024-03-06 10:53:23,409 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 10:54:49,354 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 10:55:16,373 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 2 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/607c9d_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/607c9d_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/607c9d_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/607c9d_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 11:11:23,264 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/607c9d_state.mallet.gz
2024-03-06 11:12:40,862 cisTopic INFO Model with 2 topics done!
2024-03-06 11:12:40,863 cisTopic INFO Saving model with 2 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 11:12:40,954 cisTopic INFO Running model with 5 topics
2024-03-06 11:12:40,967 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 11:14:09,509 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 11:14:37,009 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 5 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/3efb70_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/3efb70_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/3efb70_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/3efb70_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 11:31:43,881 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/3efb70_state.mallet.gz
2024-03-06 11:33:02,089 cisTopic INFO Model with 5 topics done!
2024-03-06 11:33:02,090 cisTopic INFO Saving model with 5 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 11:33:02,210 cisTopic INFO Running model with 10 topics
2024-03-06 11:33:02,224 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 11:34:30,049 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 11:34:56,968 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 10 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/56914f_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/56914f_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/56914f_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/56914f_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 11:57:16,974 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/56914f_state.mallet.gz
2024-03-06 11:58:36,725 cisTopic INFO Model with 10 topics done!
2024-03-06 11:58:36,726 cisTopic INFO Saving model with 10 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 11:58:36,873 cisTopic INFO Running model with 15 topics
2024-03-06 11:58:36,887 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 12:00:04,591 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 12:00:30,544 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 15 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/d5a078_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/d5a078_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/d5a078_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/d5a078_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 12:24:38,351 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/d5a078_state.mallet.gz
2024-03-06 12:25:59,453 cisTopic INFO Model with 15 topics done!
2024-03-06 12:25:59,454 cisTopic INFO Saving model with 15 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 12:25:59,617 cisTopic INFO Running model with 20 topics
2024-03-06 12:25:59,635 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 12:27:25,547 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 12:27:52,965 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 20 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/da4528_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/da4528_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/da4528_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/da4528_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 12:49:36,810 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/da4528_state.mallet.gz
2024-03-06 12:50:58,204 cisTopic INFO Model with 20 topics done!
2024-03-06 12:50:58,205 cisTopic INFO Saving model with 20 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 12:50:58,372 cisTopic INFO Running model with 25 topics
2024-03-06 12:50:58,386 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 12:52:25,150 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 12:52:52,157 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 25 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/c9d3ea_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/c9d3ea_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/c9d3ea_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/c9d3ea_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 13:23:17,202 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/c9d3ea_state.mallet.gz
2024-03-06 13:24:41,201 cisTopic INFO Model with 25 topics done!
2024-03-06 13:24:41,201 cisTopic INFO Saving model with 25 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 13:24:41,396 cisTopic INFO Running model with 30 topics
2024-03-06 13:24:41,414 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 13:26:08,107 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 13:26:35,899 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 30 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/9522b8_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/9522b8_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/9522b8_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/9522b8_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 13:53:23,219 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/9522b8_state.mallet.gz
2024-03-06 13:54:48,411 cisTopic INFO Model with 30 topics done!
2024-03-06 13:54:48,413 cisTopic INFO Saving model with 30 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 13:54:48,639 cisTopic INFO Running model with 35 topics
2024-03-06 13:54:48,654 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 13:56:15,826 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 13:56:44,206 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 35 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/71a4bc_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/71a4bc_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/71a4bc_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/71a4bc_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 14:24:54,558 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/71a4bc_state.mallet.gz
2024-03-06 14:26:26,621 cisTopic INFO Model with 35 topics done!
2024-03-06 14:26:26,622 cisTopic INFO Saving model with 35 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 14:26:26,894 cisTopic INFO Running model with 40 topics
2024-03-06 14:26:26,914 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 14:27:55,771 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 14:28:22,633 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 40 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/5db9d5_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/5db9d5_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/5db9d5_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/5db9d5_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 14:54:05,167 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/5db9d5_state.mallet.gz
2024-03-06 14:55:32,458 cisTopic INFO Model with 40 topics done!
2024-03-06 14:55:32,459 cisTopic INFO Saving model with 40 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 14:55:32,717 cisTopic INFO Running model with 45 topics
2024-03-06 14:55:32,732 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 14:56:59,495 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 14:57:26,628 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 45 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/cc641b_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/cc641b_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/cc641b_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/cc641b_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 15:26:12,558 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/cc641b_state.mallet.gz
2024-03-06 15:27:42,660 cisTopic INFO Model with 45 topics done!
2024-03-06 15:27:42,660 cisTopic INFO Saving model with 45 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
2024-03-06 15:27:42,926 cisTopic INFO Running model with 50 topics
2024-03-06 15:27:43,051 LDAMalletWrapper INFO Serializing temporary corpus to /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt
2024-03-06 15:29:09,831 LDAMalletWrapper INFO Converting temporary corpus to MALLET format with Mallet-202108/bin/mallet import-file --preserve-case --keep-sequence --remove-stopwords --token-regex "\S+" --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.txt --output /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet
2024-03-06 15:29:36,971 LDAMalletWrapper INFO Training MALLET LDA with Mallet-202108/bin/mallet train-topics --input /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/corpus.mallet --num-topics 50 --alpha 50 --beta 0.1 --optimize-interval 0 --num-threads 12 --output-state /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/e9e62f_state.mallet.gz --output-doc-topics /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/e9e62f_doctopics.txt --output-topic-keys /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/e9e62f_topickeys.txt --num-iterations 500 --inferencer-filename /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/e9e62f_inferencer.mallet --doc-topics-threshold 0.0 --random-seed 555
2024-03-06 16:02:19,422 LDAMalletWrapper INFO loading assigned topics from /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial/e9e62f_state.mallet.gz
2024-03-06 16:03:47,824 cisTopic INFO Model with 50 topics done!
2024-03-06 16:03:47,824 cisTopic INFO Saving model with 50 topics at /scratch/leuven/330/vsc33053/ray_spill/mallet/tutorial
[29]:
pickle.dump(
models,
open(os.path.join(out_dir, "models.pkl"), "wb")
)
Model selection
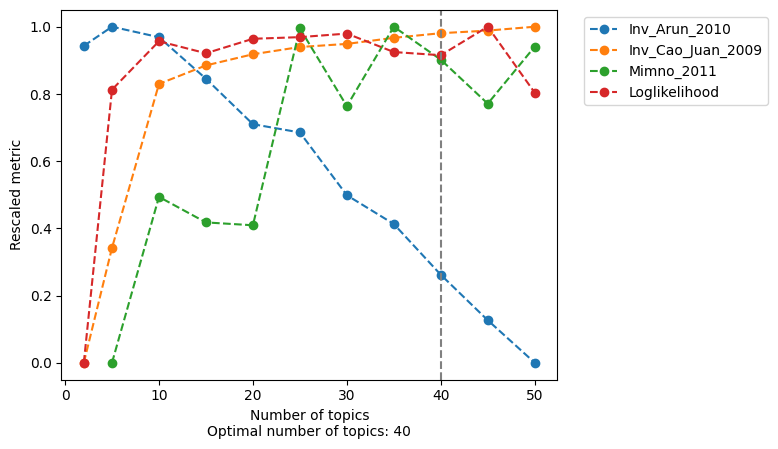
Let’s select the model with the optimal number of topics.
There is no optimal way to do this selection, however getting the exact optimal number of topics is also not critical.
To make the selection easier we implemented several metrics:
Minmo_2011: Uses the average model coherence as calculated by Mimno et al (2011). In order to reduce the impact of the number of topics, we calculate the average coherence based on the top selected average values. The better the model, the higher coherence.
Log-likelihood: Uses the log-likelihood in the last iteration as calculated by Griffiths and Steyvers (2004). The better the model, the higher the log-likelihood.
Arun_2010: Uses a density-based metric as in Arun et al (2010) using the topic-region distribution, the cell-topic distribution and the cell coverage. The better the model, the lower the metric.
Cao_Juan_2009: Uses a divergence-based metric as in Cao Juan et al (2009) using the topic-region distribution. The better the model, the lower the metric.
Note:
Note that for both the Arun and Cao metric a lower score is related to a better model, for visualization purposes we inverted these scores. In the plots below a higher score for these metrics are thus related to a better model.
Also note that not all metrics agree (see for example the Arun metric).
For scATAC-seq data models, the most helpful methods are Minmo (topic coherence) and the log-likelihood in the last iteration.
[30]:
from pycisTopic.lda_models import evaluate_models
model = evaluate_models(
models,
select_model = 40,
return_model = True
)
[31]:
cistopic_obj.add_LDA_model(model)
[32]:
pickle.dump(
cistopic_obj,
open(os.path.join(out_dir, "cistopic_obj.pkl"), "wb")
)
Clustering and visualization
We can cluster the cells (or regions) using the leiden algorithm, and perform dimensionality reductiion with UMAP and TSNE. In these examples we will focus on the cells only. For these steps, the cell-topic contributions of the model will be used.
[82]:
from pycisTopic.clust_vis import (
find_clusters,
run_umap,
run_tsne,
plot_metadata,
plot_topic,
cell_topic_heatmap
)
[93]:
find_clusters(
cistopic_obj,
target = 'cell',
k = 10,
res = [0.6, 1.2, 3],
prefix = 'pycisTopic_',
scale = True,
split_pattern = '-'
)
2024-03-06 16:40:48,444 cisTopic INFO Finding neighbours
Columns ['pycisTopic_leiden_10_0.6'] will be overwritten
Columns ['pycisTopic_leiden_10_1.2'] will be overwritten
Columns ['pycisTopic_leiden_10_3'] will be overwritten
[42]:
run_umap(
cistopic_obj,
target = 'cell', scale=True)
2024-03-06 16:21:36,432 cisTopic INFO Running UMAP
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/umap/umap_.py:1943: UserWarning: n_jobs value -1 overridden to 1 by setting random_state. Use no seed for parallelism.
warn(f"n_jobs value {self.n_jobs} overridden to 1 by setting random_state. Use no seed for parallelism.")
[43]:
run_tsne(
cistopic_obj,
target = 'cell', scale=True)
2024-03-06 16:21:56,706 cisTopic INFO Running TSNE
[70]:
plot_metadata(
cistopic_obj,
reduction_name='UMAP',
variables=['Seurat_cell_type', 'pycisTopic_leiden_10_0.6', 'pycisTopic_leiden_10_1.2', 'pycisTopic_leiden_10_3'],
target='cell', num_columns=4,
text_size=10,
dot_size=5)
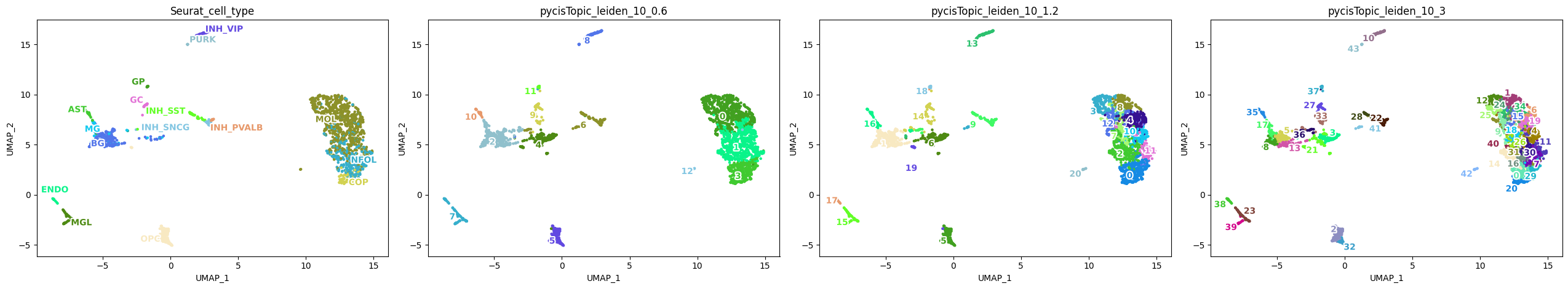
Let’s annotate each cluster based on the overlap with scRNA-seq annotations.
[94]:
annot_dict = {}
for resolution in [0.6, 1.2, 3]:
annot_dict[f"pycisTopic_leiden_10_{resolution}"] = {}
for cluster in set(cistopic_obj.cell_data[f"pycisTopic_leiden_10_{resolution}"]):
counts = cistopic_obj.cell_data.loc[
cistopic_obj.cell_data.loc[cistopic_obj.cell_data[f"pycisTopic_leiden_10_{resolution}"] == cluster].index,
"Seurat_cell_type"].value_counts()
annot_dict[f"pycisTopic_leiden_10_{resolution}"][cluster] = f"{counts.index[counts.argmax()]}({cluster})"
[95]:
annot_dict
[95]:
{'pycisTopic_leiden_10_0.6': {'11': 'GP(11)',
'6': 'INH_SST(6)',
'8': 'INH_VIP(8)',
'0': 'MOL(0)',
'7': 'MGL(7)',
'4': 'BG(4)',
'12': 'MOL(12)',
'9': 'GC(9)',
'5': 'OPC(5)',
'2': 'BG(2)',
'10': 'AST(10)',
'1': 'MOL(1)',
'3': 'NFOL(3)'},
'pycisTopic_leiden_10_1.2': {'15': 'MGL(15)',
'8': 'MOL(8)',
'12': 'MOL(12)',
'5': 'OPC(5)',
'3': 'MOL(3)',
'6': 'BG(6)',
'18': 'GP(18)',
'14': 'GC(14)',
'19': 'OPC(19)',
'20': 'MOL(20)',
'17': 'ENDO(17)',
'16': 'AST(16)',
'2': 'MOL(2)',
'10': 'MOL(10)',
'1': 'BG(1)',
'11': 'MOL(11)',
'0': 'NFOL(0)',
'4': 'MOL(4)',
'9': 'INH_SST(9)',
'13': 'INH_VIP(13)',
'7': 'MOL(7)'},
'pycisTopic_leiden_10_3': {'21': 'OPC(21)',
'31': 'MOL(31)',
'15': 'MOL(15)',
'8': 'BG(8)',
'37': 'GP(37)',
'12': 'MOL(12)',
'41': 'INH_SST(41)',
'5': 'BG(5)',
'27': 'GC(27)',
'43': 'PURK(43)',
'6': 'MOL(6)',
'3': 'BG(3)',
'38': 'ENDO(38)',
'18': 'MOL(18)',
'14': 'NFOL(14)',
'19': 'MOL(19)',
'20': 'COP(20)',
'30': 'MOL(30)',
'17': 'BG(17)',
'34': 'MOL(34)',
'25': 'MOL(25)',
'39': 'MGL(39)',
'42': 'MOL(42)',
'33': 'GC(33)',
'24': 'MOL(24)',
'28': 'INH_SST(28)',
'29': 'NFOL(29)',
'16': 'NFOL(16)',
'2': 'OPC(2)',
'10': 'INH_VIP(10)',
'1': 'MOL(1)',
'11': 'MOL(11)',
'22': 'INH_SNCG(22)',
'26': 'MOL(26)',
'35': 'AST(35)',
'23': 'MGL(23)',
'36': 'MG(36)',
'0': 'NFOL(0)',
'40': 'MOL(40)',
'4': 'MOL(4)',
'9': 'MOL(9)',
'13': 'BG(13)',
'7': 'NFOL(7)',
'32': 'OPC(32)'}}
[96]:
for resolution in [0.6, 1.2, 3]:
cistopic_obj.cell_data[f'pycisTopic_leiden_10_{resolution}'] = [
annot_dict[f'pycisTopic_leiden_10_{resolution}'][x] for x in cistopic_obj.cell_data[f'pycisTopic_leiden_10_{resolution}'].tolist()
]
[97]:
plot_metadata(
cistopic_obj,
reduction_name='UMAP',
variables=['Seurat_cell_type', 'pycisTopic_leiden_10_0.6', 'pycisTopic_leiden_10_1.2', 'pycisTopic_leiden_10_3'],
target='cell', num_columns=4,
text_size=10,
dot_size=5)
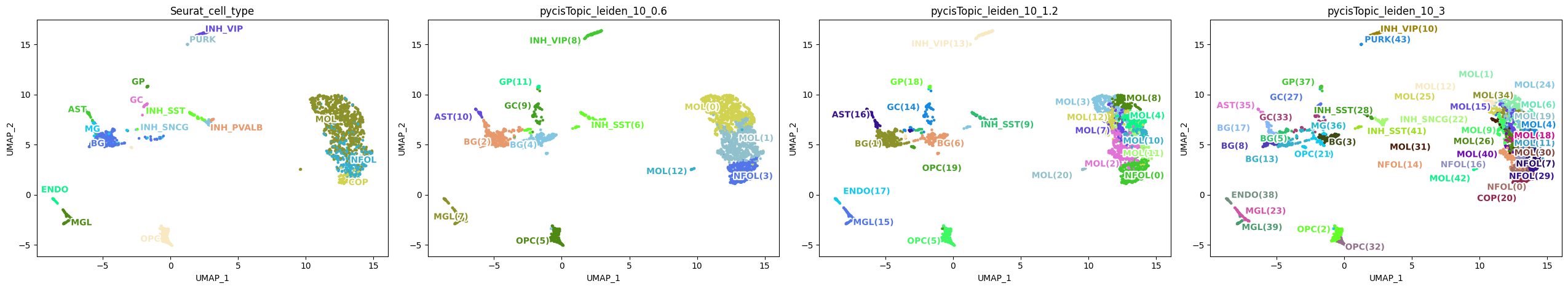
We can also plot continuous values.
[78]:
plot_metadata(
cistopic_obj,
reduction_name='UMAP',
variables=['log10_unique_fragments_count', 'tss_enrichment', 'Doublet_scores_fragments', 'fraction_of_fragments_in_peaks'],
target='cell', num_columns=4,
text_size=10,
dot_size=5)
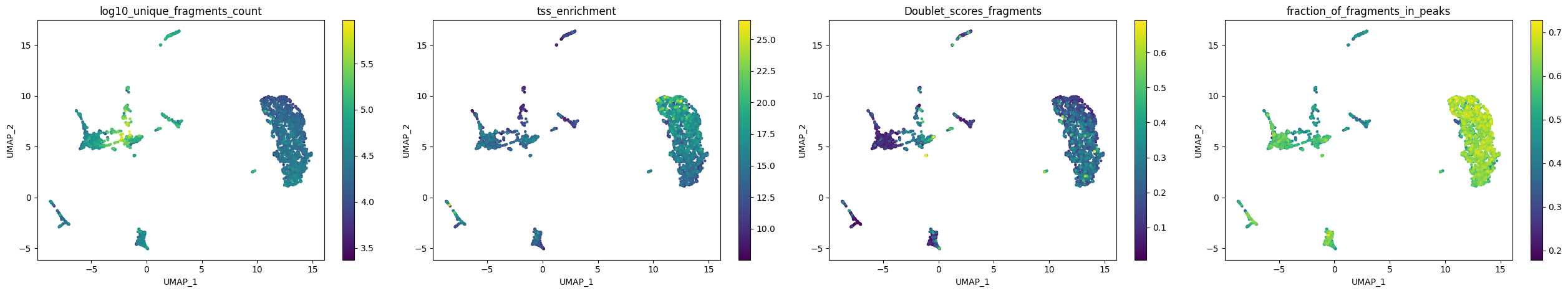
Let’s visualize the cell-topic contributions.
[81]:
plot_topic(
cistopic_obj,
reduction_name = 'UMAP',
target = 'cell',
num_columns=5
)
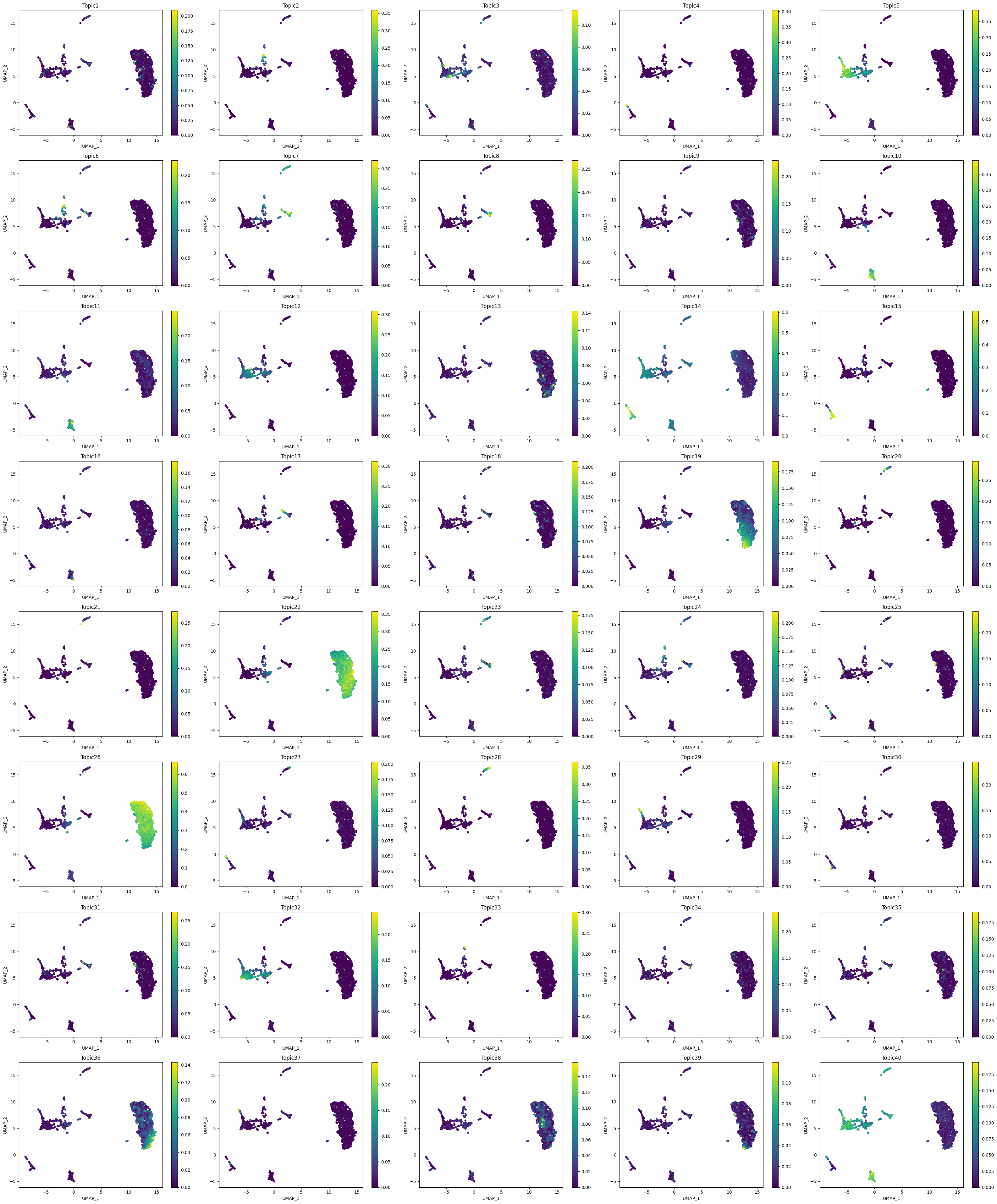
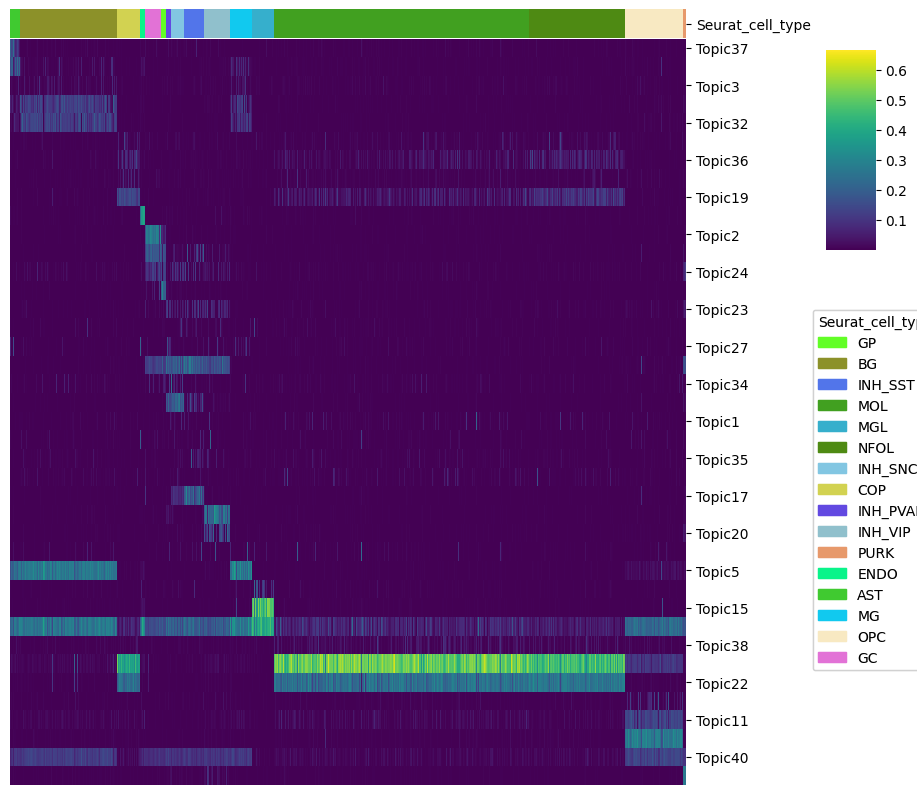
Or we can also draw a heatmap with the topic contributions (and annotations).
[90]:
cell_topic_heatmap(
cistopic_obj,
variables = ['Seurat_cell_type'],
scale = False,
legend_loc_x = 1.0,
legend_loc_y = -1.2,
legend_dist_y = -1,
figsize = (10, 10)
)
Topic binarization & QC
Next we can binarize topic-region and cell-topic distributions. The first is useful for exploring the topics with other tools that work with region sets (e.g. GREAT, cisTarget); while the latter is useful to automatically annotate topics.
We will first binarize the topic-region distributions. There are several methods that can be used for this: ‘otsu’ (Otsu, 1979), ‘yen’ (Yen et al., 1995), ‘li’ (Li & Lee, 1993), ‘aucell’ (Van de Sande et al., 2020) or ‘ntop’ (Taking the top n regions per topic). Otsu and Yen’s methods work well for topic-region distributions; however for some downstream analyses (e.g. deep learning) it may be convenient to use ntop to have balanced region sets.
[103]:
from pycisTopic.topic_binarization import binarize_topics
[116]:
region_bin_topics_top_3k = binarize_topics(
cistopic_obj, method='ntop', ntop = 3_000,
plot=True, num_columns=5
)

[117]:
region_bin_topics_otsu = binarize_topics(
cistopic_obj, method='otsu',
plot=True, num_columns=5
)

Similarly, we can now binarize the cell-topic distribions.
[101]:
binarized_cell_topic = binarize_topics(
cistopic_obj,
target='cell',
method='li',
plot=True,
num_columns=5, nbins=100)
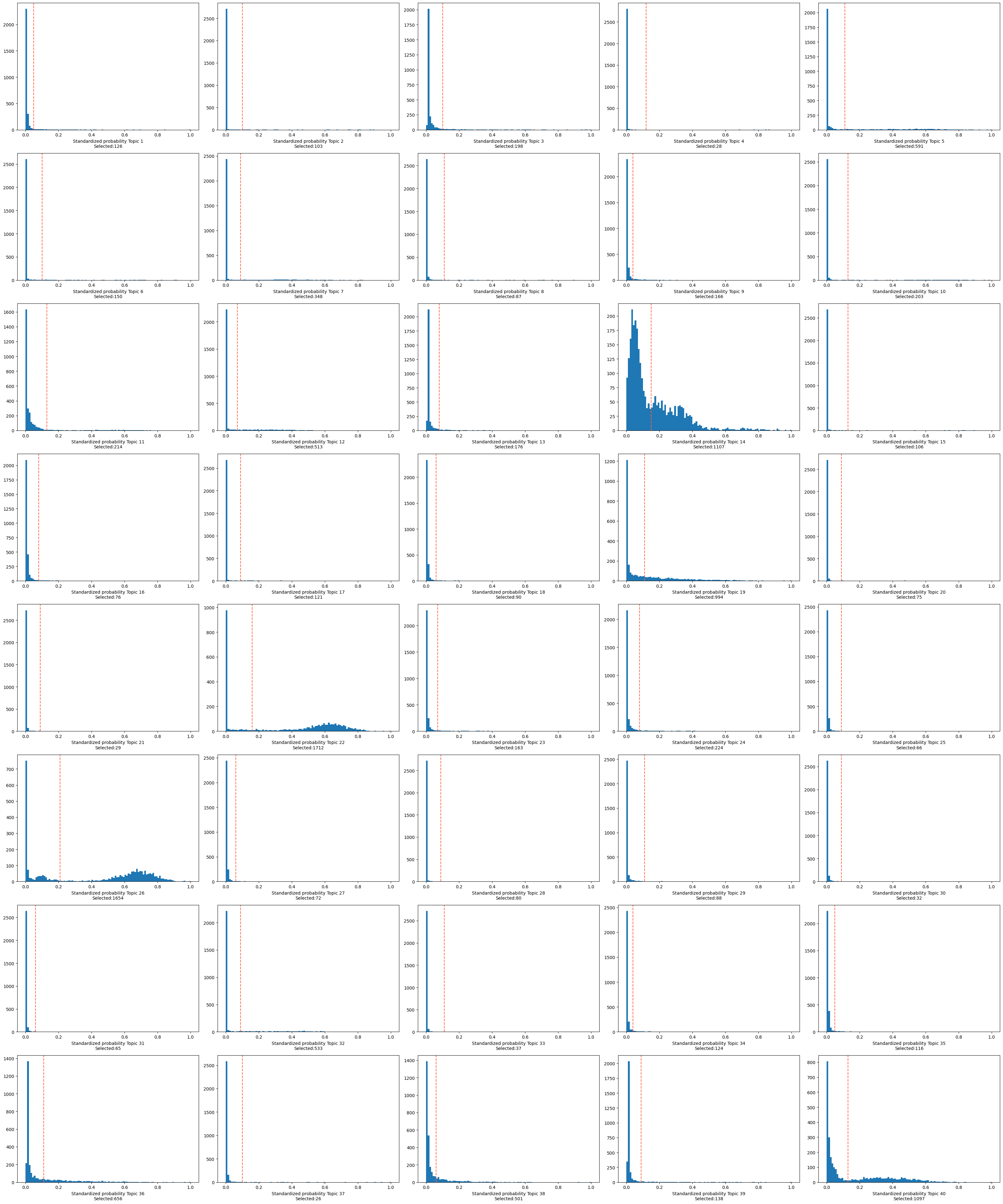
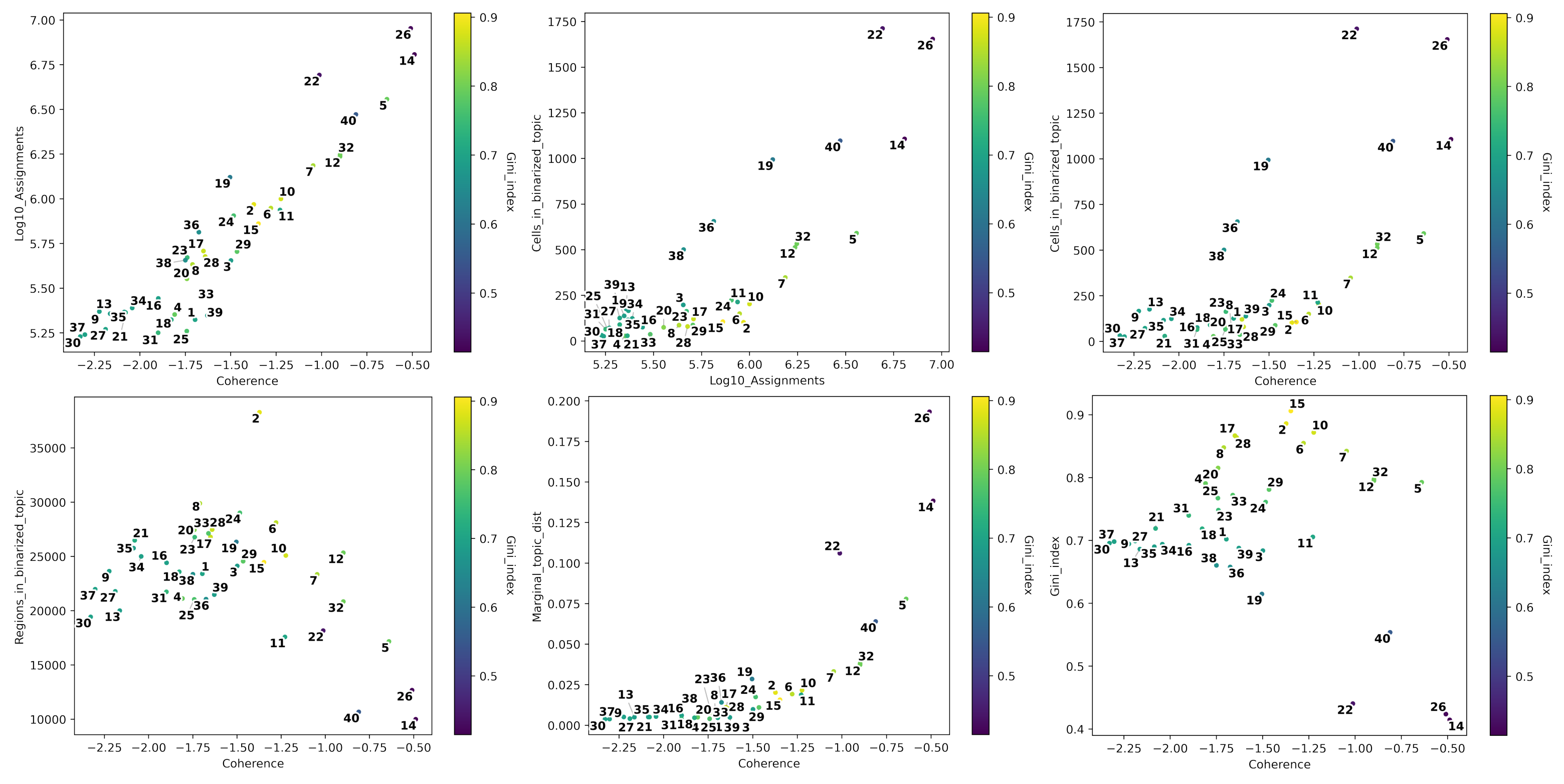
Following, we can compute the topic quality control metrics. These include:
Number of assignments
Topic coherence (Mimno et al., 2011): Measures to which extent high scoring regions in the topic are actually co-accessible in the original data. If it is low it indicates that the topic is rather random. The higher, the better is a topic.
The marginal topic distribution: Indicates how much each topic contributes to the model. The higher, the better is a topic.
The gini index: Value between 0 and 1, that indicates the specificity of topics (0: General, 1:Specific)
If topics have been binarized, the number of regions/cells per topic will be added.
[121]:
from pycisTopic.topic_qc import compute_topic_metrics, plot_topic_qc, topic_annotation
import matplotlib.pyplot as plt
from pycisTopic.utils import fig2img
[118]:
topic_qc_metrics = compute_topic_metrics(cistopic_obj)
[119]:
fig_dict={}
fig_dict['CoherenceVSAssignments']=plot_topic_qc(topic_qc_metrics, var_x='Coherence', var_y='Log10_Assignments', var_color='Gini_index', plot=False, return_fig=True)
fig_dict['AssignmentsVSCells_in_bin']=plot_topic_qc(topic_qc_metrics, var_x='Log10_Assignments', var_y='Cells_in_binarized_topic', var_color='Gini_index', plot=False, return_fig=True)
fig_dict['CoherenceVSCells_in_bin']=plot_topic_qc(topic_qc_metrics, var_x='Coherence', var_y='Cells_in_binarized_topic', var_color='Gini_index', plot=False, return_fig=True)
fig_dict['CoherenceVSRegions_in_bin']=plot_topic_qc(topic_qc_metrics, var_x='Coherence', var_y='Regions_in_binarized_topic', var_color='Gini_index', plot=False, return_fig=True)
fig_dict['CoherenceVSMarginal_dist']=plot_topic_qc(topic_qc_metrics, var_x='Coherence', var_y='Marginal_topic_dist', var_color='Gini_index', plot=False, return_fig=True)
fig_dict['CoherenceVSGini_index']=plot_topic_qc(topic_qc_metrics, var_x='Coherence', var_y='Gini_index', var_color='Gini_index', plot=False, return_fig=True)
[120]:
# Plot topic stats in one figure
fig=plt.figure(figsize=(40, 43))
i = 1
for fig_ in fig_dict.keys():
plt.subplot(2, 3, i)
img = fig2img(fig_dict[fig_]) #To convert figures to png to plot together, see .utils.py. This converts the figure to png.
plt.imshow(img)
plt.axis('off')
i += 1
plt.subplots_adjust(wspace=0, hspace=-0.70)
plt.show()
Next, we can automatically annotate topics, in this case by cell type. Here we calculate the proportion of cells in each group that are assigned to the binarized topic in comparison to the ratio in the whole data set. We will consider a topic as general if the difference between the ration of cells in the whole data set in the binarized topic and the ratio of total cells in the assigned groups is above 0.2. This indicates that the topic is general, and the propotion test may fail if the topic is enriched in both foreground (the group) and background (the whole data set); resulting in a big difference between the ratios.
[122]:
topic_annot = topic_annotation(
cistopic_obj,
annot_var='Seurat_cell_type',
binarized_cell_topic=binarized_cell_topic,
general_topic_thr = 0.2
)
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/statsmodels/stats/weightstats.py:792: RuntimeWarning: divide by zero encountered in scalar divide
zstat = value / std
[123]:
topic_annot
[123]:
| Seurat_cell_type | Ratio_cells_in_topic | Ratio_group_in_population | is_general | |
|---|---|---|---|---|
| Topic1 | MOL, INH_SNCG | 0.044164 | 0.274448 | False |
| Topic2 | GP, GC | 0.036102 | 0.021381 | False |
| Topic3 | BG, MG | 0.069401 | 0.121276 | False |
| Topic4 | ENDO | 0.009814 | 0.005608 | False |
| Topic5 | BG, AST, MG | 0.20715 | 0.131791 | False |
| Topic6 | INH_SST, GP, GC | 0.052576 | 0.04171 | False |
| Topic7 | INH_SST, GP, INH_SNCG, INH_VIP, PURK, INH_PVAL... | 0.121977 | 0.090782 | False |
| Topic8 | INH_SST, INH_SNCG, INH_PVALB | 0.030494 | 0.038906 | False |
| Topic9 | MOL, GC | 0.058184 | 0.276902 | False |
| Topic10 | OPC | 0.071153 | 0.059586 | False |
| Topic11 | OPC | 0.075009 | 0.059586 | False |
| Topic12 | BG, MG | 0.179811 | 0.121276 | False |
| Topic13 | NFOL, COP | 0.061689 | 0.120925 | False |
| Topic14 | INH_SST, BG, GP, MGL, INH_SNCG, INH_VIP, PURK,... | 0.388013 | 0.31055 | False |
| Topic15 | MGL | 0.037154 | 0.022783 | False |
| Topic16 | OPC | 0.026639 | 0.059586 | False |
| Topic17 | INH_SST, INH_SNCG | 0.042411 | 0.033649 | False |
| Topic18 | INH_SST, INH_VIP | 0.031546 | 0.047319 | False |
| Topic19 | MOL, NFOL, COP | 0.348405 | 0.382054 | False |
| Topic20 | INH_VIP, PURK | 0.026288 | 0.030494 | False |
| Topic21 | INH_VIP, PURK | 0.010165 | 0.030494 | False |
| Topic22 | MOL, NFOL, COP | 0.60007 | 0.382054 | True |
| Topic23 | INH_SST, INH_SNCG, INH_VIP, PURK, INH_PVALB | 0.057133 | 0.069401 | False |
| Topic24 | INH_SST, GP, INH_SNCG, INH_VIP, PURK, GC | 0.078514 | 0.085524 | False |
| Topic25 | INH_SST, INH_SNCG, MG | 0.023134 | 0.055731 | False |
| Topic26 | MOL, NFOL, COP | 0.579741 | 0.382054 | False |
| Topic27 | INH_VIP, INH_PVALB, ENDO, MG | 0.025237 | 0.059937 | False |
| Topic28 | INH_VIP | 0.028041 | 0.026989 | False |
| Topic29 | BG, AST, MG | 0.030845 | 0.131791 | False |
| Topic30 | MGL | 0.011216 | 0.022783 | False |
| Topic31 | INH_SST, INH_SNCG, INH_PVALB | 0.022783 | 0.038906 | False |
| Topic32 | BG, AST, MG | 0.186821 | 0.131791 | False |
| Topic33 | GP, GC | 0.012969 | 0.021381 | False |
| Topic34 | BG, GP, INH_SNCG, GC | 0.043463 | 0.133894 | False |
| Topic35 | INH_SST, INH_SNCG, INH_VIP | 0.040659 | 0.060638 | False |
| Topic36 | NFOL, COP | 0.229933 | 0.120925 | False |
| Topic37 | AST | 0.009113 | 0.010515 | False |
| Topic38 | MOL, NFOL | 0.175605 | 0.35892 | False |
| Topic39 | NFOL, COP | 0.04837 | 0.120925 | False |
| Topic40 | INH_SST, BG, GP, INH_SNCG, INH_VIP, PURK, INH_... | 0.384508 | 0.287767 | False |
Differentially Accessible Regions (DARs)
Together with working with regulatory topics, we can also identify differentially accessible regions (DARs) between cell types. First, we will impute the region accessibility exploting the cell-topic and topic-region probabilities. To shrink very low probability values to 0, we use a scale factor (by default: 10^6).
[131]:
from pycisTopic.diff_features import (
impute_accessibility,
normalize_scores,
find_highly_variable_features,
find_diff_features
)
import numpy as np
[125]:
imputed_acc_obj = impute_accessibility(
cistopic_obj,
selected_cells=None,
selected_regions=None,
scale_factor=10**6
)
2024-03-06 16:55:01,494 cisTopic INFO Imputing region accessibility
2024-03-06 16:55:01,494 cisTopic INFO Impute region accessibility for regions 0-20000
2024-03-06 16:55:01,927 cisTopic INFO Impute region accessibility for regions 20000-40000
2024-03-06 16:55:02,201 cisTopic INFO Impute region accessibility for regions 40000-60000
2024-03-06 16:55:02,472 cisTopic INFO Impute region accessibility for regions 60000-80000
2024-03-06 16:55:02,746 cisTopic INFO Impute region accessibility for regions 80000-100000
2024-03-06 16:55:03,017 cisTopic INFO Impute region accessibility for regions 100000-120000
2024-03-06 16:55:03,291 cisTopic INFO Impute region accessibility for regions 120000-140000
2024-03-06 16:55:03,564 cisTopic INFO Impute region accessibility for regions 140000-160000
2024-03-06 16:55:03,836 cisTopic INFO Impute region accessibility for regions 160000-180000
2024-03-06 16:55:04,104 cisTopic INFO Impute region accessibility for regions 180000-200000
2024-03-06 16:55:04,373 cisTopic INFO Impute region accessibility for regions 200000-220000
2024-03-06 16:55:04,642 cisTopic INFO Impute region accessibility for regions 220000-240000
2024-03-06 16:55:04,911 cisTopic INFO Impute region accessibility for regions 240000-260000
2024-03-06 16:55:05,178 cisTopic INFO Impute region accessibility for regions 260000-280000
2024-03-06 16:55:05,458 cisTopic INFO Impute region accessibility for regions 280000-300000
2024-03-06 16:55:05,725 cisTopic INFO Impute region accessibility for regions 300000-320000
2024-03-06 16:55:05,995 cisTopic INFO Impute region accessibility for regions 320000-340000
2024-03-06 16:55:06,263 cisTopic INFO Impute region accessibility for regions 340000-360000
2024-03-06 16:55:06,535 cisTopic INFO Impute region accessibility for regions 360000-380000
2024-03-06 16:55:06,803 cisTopic INFO Impute region accessibility for regions 380000-400000
2024-03-06 16:55:07,071 cisTopic INFO Impute region accessibility for regions 400000-420000
2024-03-06 16:55:07,339 cisTopic INFO Impute region accessibility for regions 420000-440000
2024-03-06 16:55:07,578 cisTopic INFO Done!
[126]:
normalized_imputed_acc_obj = normalize_scores(imputed_acc_obj, scale_factor=10**4)
2024-03-06 16:55:12,062 cisTopic INFO Normalizing imputed data
2024-03-06 16:55:18,978 cisTopic INFO Done!
Optionally, we can identify highly variable regions. This is not mandatory, but will speed up the hypothesis testing step for identifying DARs.
[129]:
variable_regions = find_highly_variable_features(
normalized_imputed_acc_obj,
min_disp = 0.05,
min_mean = 0.0125,
max_mean = 3,
max_disp = np.inf,
n_bins=20,
n_top_features=None,
plot=True
)
2024-03-06 16:55:41,694 cisTopic INFO Calculating mean
2024-03-06 16:55:43,121 cisTopic INFO Calculating variance
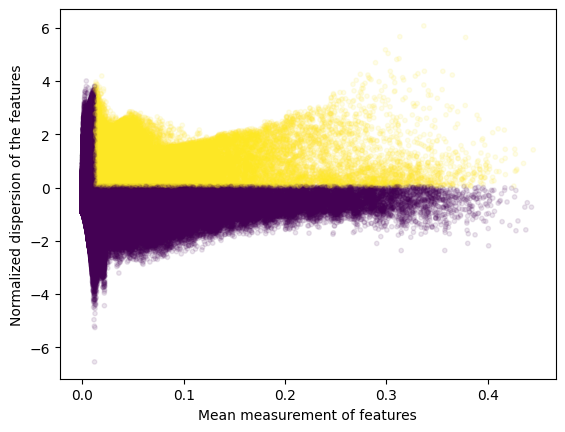
2024-03-06 16:56:02,058 cisTopic INFO Done!
[130]:
len(variable_regions)
[130]:
59709
We can now identify differentially accessible regions between groups. By default, this function will perform a Wilcoxon rank-sum test between each group using the specified variable and the rest. Alternatively, specified contrast can be provided as a list with foreground and background groups (e.g. for group 1 versus group 2 and 3, and group 2 versus group 1 and 3: [[[‘Group_1’], [‘Group_2, ‘Group_3’]], [[‘Group_2’], [‘Group_1, ‘Group_3’]]]).
[132]:
markers_dict= find_diff_features(
cistopic_obj,
imputed_acc_obj,
variable='Seurat_cell_type',
var_features=variable_regions,
contrasts=None,
adjpval_thr=0.05,
log2fc_thr=np.log2(1.5),
n_cpu=5,
_temp_dir='/scratch/leuven/330/vsc33053/ray_spill',
split_pattern = '-'
)
2024-03-06 16:57:06,033 INFO worker.py:1724 -- Started a local Ray instance.
2024-03-06 16:57:06,739 cisTopic INFO Subsetting data for AST (30 of 2853)
2024-03-06 16:57:10,317 cisTopic INFO Computing p-value for AST
2024-03-06 16:57:23,252 cisTopic INFO Computing log2FC for AST
2024-03-06 16:57:24,824 cisTopic INFO AST done!
2024-03-06 16:57:24,827 cisTopic INFO Subsetting data for BG (283 of 2853)
2024-03-06 16:57:24,875 cisTopic INFO Computing p-value for BG
2024-03-06 16:57:30,629 cisTopic INFO Computing log2FC for BG
2024-03-06 16:57:30,667 cisTopic INFO BG done!
2024-03-06 16:57:30,669 cisTopic INFO Subsetting data for COP (66 of 2853)
2024-03-06 16:57:30,700 cisTopic INFO Computing p-value for COP
2024-03-06 16:57:36,519 cisTopic INFO Computing log2FC for COP
2024-03-06 16:57:36,552 cisTopic INFO COP done!
2024-03-06 16:57:36,554 cisTopic INFO Subsetting data for ENDO (16 of 2853)
2024-03-06 16:57:36,582 cisTopic INFO Computing p-value for ENDO
2024-03-06 16:57:42,393 cisTopic INFO Computing log2FC for ENDO
2024-03-06 16:57:42,423 cisTopic INFO ENDO done!
2024-03-06 16:57:42,425 cisTopic INFO Subsetting data for GC (45 of 2853)
2024-03-06 16:57:42,459 cisTopic INFO Computing p-value for GC
2024-03-06 16:57:48,248 cisTopic INFO Computing log2FC for GC
2024-03-06 16:57:48,283 cisTopic INFO GC done!
2024-03-06 16:57:48,285 cisTopic INFO Subsetting data for GP (16 of 2853)
2024-03-06 16:57:48,311 cisTopic INFO Computing p-value for GP
2024-03-06 16:57:54,105 cisTopic INFO Computing log2FC for GP
2024-03-06 16:57:54,140 cisTopic INFO GP done!
2024-03-06 16:57:54,142 cisTopic INFO Subsetting data for INH_PVALB (15 of 2853)
2024-03-06 16:57:54,169 cisTopic INFO Computing p-value for INH_PVALB
2024-03-06 16:58:00,011 cisTopic INFO Computing log2FC for INH_PVALB
2024-03-06 16:58:00,045 cisTopic INFO INH_PVALB done!
2024-03-06 16:58:00,047 cisTopic INFO Subsetting data for INH_SNCG (38 of 2853)
2024-03-06 16:58:00,094 cisTopic INFO Computing p-value for INH_SNCG
2024-03-06 16:58:06,018 cisTopic INFO Computing log2FC for INH_SNCG
2024-03-06 16:58:06,054 cisTopic INFO INH_SNCG done!
2024-03-06 16:58:06,056 cisTopic INFO Subsetting data for INH_SST (58 of 2853)
2024-03-06 16:58:06,107 cisTopic INFO Computing p-value for INH_SST
2024-03-06 16:58:11,934 cisTopic INFO Computing log2FC for INH_SST
2024-03-06 16:58:11,968 cisTopic INFO INH_SST done!
2024-03-06 16:58:11,970 cisTopic INFO Subsetting data for INH_VIP (77 of 2853)
2024-03-06 16:58:12,011 cisTopic INFO Computing p-value for INH_VIP
2024-03-06 16:58:17,978 cisTopic INFO Computing log2FC for INH_VIP
2024-03-06 16:58:18,021 cisTopic INFO INH_VIP done!
2024-03-06 16:58:18,023 cisTopic INFO Subsetting data for MG (63 of 2853)
2024-03-06 16:58:18,052 cisTopic INFO Computing p-value for MG
2024-03-06 16:58:24,203 cisTopic INFO Computing log2FC for MG
2024-03-06 16:58:24,239 cisTopic INFO MG done!
2024-03-06 16:58:24,241 cisTopic INFO Subsetting data for MGL (65 of 2853)
2024-03-06 16:58:24,272 cisTopic INFO Computing p-value for MGL
2024-03-06 16:58:30,122 cisTopic INFO Computing log2FC for MGL
2024-03-06 16:58:30,154 cisTopic INFO MGL done!
2024-03-06 16:58:30,157 cisTopic INFO Subsetting data for MOL (745 of 2853)
2024-03-06 16:58:30,191 cisTopic INFO Computing p-value for MOL
2024-03-06 16:58:35,955 cisTopic INFO Computing log2FC for MOL
2024-03-06 16:58:35,996 cisTopic INFO MOL done!
2024-03-06 16:58:35,998 cisTopic INFO Subsetting data for NFOL (279 of 2853)
2024-03-06 16:58:36,062 cisTopic INFO Computing p-value for NFOL
2024-03-06 16:58:41,890 cisTopic INFO Computing log2FC for NFOL
2024-03-06 16:58:41,925 cisTopic INFO NFOL done!
2024-03-06 16:58:41,927 cisTopic INFO Subsetting data for OPC (170 of 2853)
2024-03-06 16:58:41,959 cisTopic INFO Computing p-value for OPC
2024-03-06 16:58:47,808 cisTopic INFO Computing log2FC for OPC
2024-03-06 16:58:47,842 cisTopic INFO OPC done!
2024-03-06 16:58:47,844 cisTopic INFO Subsetting data for PURK (10 of 2853)
2024-03-06 16:58:47,870 cisTopic INFO Computing p-value for PURK
2024-03-06 16:58:53,610 cisTopic INFO Computing log2FC for PURK
2024-03-06 16:58:53,639 cisTopic INFO PURK done!
We can also plot region accessibility onto the cell-topic UMAP. For example, let’s check how the best DARs for some cell types look like.
[133]:
from pycisTopic.clust_vis import plot_imputed_features
[134]:
plot_imputed_features(
cistopic_obj,
reduction_name='UMAP',
imputed_data=imputed_acc_obj,
features=[markers_dict[x].index.tolist()[0] for x in ['BG', 'GC', 'INH_SST', 'COP']],
scale=False,
num_columns=4
)
[139]:
print("Number of DARs found:")
print("---------------------")
for x in markers_dict:
print(f" {x}: {len(markers_dict[x])}")
Number of DARs found:
---------------------
AST: 22352
BG: 26552
COP: 11739
ENDO: 9340
GC: 12518
GP: 12200
INH_PVALB: 12405
INH_SNCG: 12536
INH_SST: 12082
INH_VIP: 14240
MG: 25522
MGL: 9438
MOL: 20705
NFOL: 16428
OPC: 17949
PURK: 13188
Save region sets
[142]:
os.makedirs(os.path.join(out_dir, "region_sets"), exist_ok = True)
os.makedirs(os.path.join(out_dir, "region_sets", "Topics_otsu"), exist_ok = True)
os.makedirs(os.path.join(out_dir, "region_sets", "Topics_top_3k"), exist_ok = True)
os.makedirs(os.path.join(out_dir, "region_sets", "DARs_cell_type"), exist_ok = True)
[141]:
from pycisTopic.utils import region_names_to_coordinates
[143]:
for topic in region_bin_topics_otsu:
region_names_to_coordinates(
region_bin_topics_otsu[topic].index
).sort_values(
["Chromosome", "Start", "End"]
).to_csv(
os.path.join(out_dir, "region_sets", "Topics_otsu", f"{topic}.bed"),
sep = "\t",
header = False, index = False
)
[144]:
for topic in region_bin_topics_top_3k:
region_names_to_coordinates(
region_bin_topics_top_3k[topic].index
).sort_values(
["Chromosome", "Start", "End"]
).to_csv(
os.path.join(out_dir, "region_sets", "Topics_top_3k", f"{topic}.bed"),
sep = "\t",
header = False, index = False
)
[145]:
for cell_type in markers_dict:
region_names_to_coordinates(
markers_dict[cell_type].index
).sort_values(
["Chromosome", "Start", "End"]
).to_csv(
os.path.join(out_dir, "region_sets", "DARs_cell_type", f"{cell_type}.bed"),
sep = "\t",
header = False, index = False
)
Gene activity
Now we can infer gene activity.
In this function there are several options to evaluate: - Search space: The user can choose whether the search space should be include other genes or not (use_gene_boundaries), and the minimum and maximum distance it should have (upstream and downstream) - Distance weight: The parameters related to the distance weight measure the impact of distance when inferring region to gene weights as an exponential function. The user can control whether this weight should be used (distance_weight) and the effect of the distance (decay_rate). - Gene size weight: Large genes may have more peaks by chance. The user can optionally apply a weight based on the size of each gene (gene_size_weight), which by default is dividing the size of each gene by the median gene size in the genome. Alternatively, the user can also use average_scores which will calculate the gene activity as the mean weighted region accessibility of all regions linked to the gene. - Gini weight: This weight will give more importance to specific regions (gini_weight).
[157]:
import pyranges as pr
from pycisTopic.gene_activity import get_gene_activity
[150]:
chromsizes = pd.read_table(os.path.join(out_dir, "qc", "hg38.chrom_sizes_and_alias.tsv"))
chromsizes
[150]:
| # ucsc | length | ensembl | refseq_id | genbank_id | |
|---|---|---|---|---|---|
| 0 | chr1 | 248956422 | 1 | NC_000001.11 | CM000663.2 |
| 1 | chr2 | 242193529 | 2 | NC_000002.12 | CM000664.2 |
| 2 | chr3 | 198295559 | 3 | NC_000003.12 | CM000665.2 |
| 3 | chr4 | 190214555 | 4 | NC_000004.12 | CM000666.2 |
| 4 | chr5 | 181538259 | 5 | NC_000005.10 | CM000667.2 |
| ... | ... | ... | ... | ... | ... |
| 449 | chrUn_KI270539v1 | 993 | HSCHRUN_RANDOM_146 | NT_187442.1 | KI270539.1 |
| 450 | chrUn_KI270385v1 | 990 | HSCHRUN_RANDOM_195 | NT_187487.1 | KI270385.1 |
| 451 | chrUn_KI270423v1 | 981 | HSCHRUN_RANDOM_121 | NT_187417.1 | KI270423.1 |
| 452 | chrUn_KI270392v1 | 971 | HSCHRUN_RANDOM_193 | NT_187485.1 | KI270392.1 |
| 453 | chrUn_KI270394v1 | 970 | HSCHRUN_RANDOM_187 | NT_187479.1 | KI270394.1 |
454 rows × 5 columns
[153]:
chromsizes.rename({"# ucsc": "Chromosome", "length": "End"}, axis = 1, inplace = True)
chromsizes["Start"] = 0
chromsizes = pr.PyRanges(chromsizes[["Chromosome", "Start", "End"]])
[154]:
chromsizes
[154]:
| Chromosome | Start | End | |
|---|---|---|---|
| 0 | chr1 | 0 | 248956422 |
| 1 | chr1_GL383518v1_alt | 0 | 182439 |
| 2 | chr1_GL383519v1_alt | 0 | 110268 |
| 3 | chr1_GL383520v2_alt | 0 | 366580 |
| 4 | chr1_KI270706v1_random | 0 | 175055 |
| ... | ... | ... | ... |
| 449 | chrX_KI270880v1_alt | 0 | 284869 |
| 450 | chrX_KI270881v1_alt | 0 | 144206 |
| 451 | chrX_KI270913v1_alt | 0 | 274009 |
| 452 | chrY | 0 | 57227415 |
| 453 | chrY_KI270740v1_random | 0 | 37240 |
454 rows × 3 columns
[167]:
pr_annotation = pd.read_table(
os.path.join(out_dir, "qc", "tss.bed")
).rename(
{"Name": "Gene", "# Chromosome": "Chromosome"}, axis = 1)
pr_annotation["Transcription_Start_Site"] = pr_annotation["Start"]
pr_annotation = pr.PyRanges(pr_annotation)
pr_annotation
[167]:
| Chromosome | Start | End | Gene | Score | Strand | Transcript_type | Transcription_Start_Site | |
|---|---|---|---|---|---|---|---|---|
| 0 | GL000009.2 | 58375 | 58376 | NaN | . | - | protein_coding | 58375 |
| 1 | GL000194.1 | 115017 | 115018 | NaN | . | - | protein_coding | 115017 |
| 2 | GL000194.1 | 115054 | 115055 | MAFIP | . | - | protein_coding | 115054 |
| 3 | GL000195.1 | 49163 | 49164 | NaN | . | - | protein_coding | 49163 |
| 4 | GL000213.1 | 139654 | 139655 | NaN | . | - | protein_coding | 139654 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 87275 | chrY | 2500554 | 2500555 | ZBED1 | . | - | protein_coding | 2500554 |
| 87276 | chrY | 2500894 | 2500895 | ZBED1 | . | - | protein_coding | 2500894 |
| 87277 | chrY | 318795 | 318796 | GTPBP6 | . | - | protein_coding | 318795 |
| 87278 | chrY | 1212648 | 1212649 | CRLF2 | . | - | protein_coding | 1212648 |
| 87279 | chrY | 1212633 | 1212634 | CRLF2 | . | - | protein_coding | 1212633 |
87280 rows × 8 columns
[168]:
gene_act, weigths = get_gene_activity(
imputed_acc_obj,
pr_annotation,
chromsizes,
use_gene_boundaries=True, # Whether to use the whole search space or stop when encountering another gene
upstream=[1000, 100000], # Search space upstream. The minimum means that even if there is a gene right next to it
# these bp will be taken (1kbp here)
downstream=[1000,100000], # Search space downstream
distance_weight=True, # Whether to add a distance weight (an exponential function, the weight will decrease with distance)
decay_rate=1, # Exponent for the distance exponential funciton (the higher the faster will be the decrease)
extend_gene_body_upstream=10000, # Number of bp upstream immune to the distance weight (their value will be maximum for
#this weight)
extend_gene_body_downstream=500, # Number of bp downstream immune to the distance weight
gene_size_weight=False, # Whether to add a weights based on the length of the gene
gene_size_scale_factor='median', # Dividend to calculate the gene size weigth. Default is the median value of all genes
#in the genome
remove_promoters=False, # Whether to remove promoters when computing gene activity scores
average_scores=True, # Whether to divide by the total number of region assigned to a gene when calculating the gene
#activity score
scale_factor=1, # Value to multiply for the final gene activity matrix
extend_tss=[10,10], # Space to consider a promoter
gini_weight = True, # Whether to add a gini index weigth. The more unique the region is, the higher this weight will be
return_weights= True, # Whether to return the final weights
project='Gene_activity') # Project name for the gene activity object
Warning! Start and End columns now have different dtypes: int32 and int64
Warning! Start and End columns now have different dtypes: int32 and int64
2024-03-06 17:13:02,671 cisTopic INFO Calculating gene boundaries
Warning! Start and End columns now have different dtypes: int32 and int64
Warning! Start and End columns now have different dtypes: int32 and int64
Warning! Start and End columns now have different dtypes: int32 and int64
2024-03-06 17:13:17,782 cisTopic INFO Calculating distances
join: Strand data from other will be added as strand data to self.
If this is undesired use the flag apply_strand_suffix=False.
To turn off the warning set apply_strand_suffix to True or False.
2024-03-06 17:13:33,422 cisTopic INFO Calculating distance weigths
2024-03-06 17:13:34,066 cisTopic INFO Distance weights done
2024-03-06 17:13:34,066 cisTopic INFO Calculating gini weights
2024-03-06 17:13:47,195 cisTopic INFO Getting gene activity scores
2024-03-06 17:17:14,591 cisTopic INFO Creating imputed features object
As we did before for the imputed region accessibility, we can also infer the Differentially Accessible Genes (DAGs).
[169]:
DAG_markers_dict= find_diff_features(
cistopic_obj,
gene_act,
variable='Seurat_cell_type',
var_features=None,
contrasts=None,
adjpval_thr=0.05,
log2fc_thr=np.log2(1.5),
n_cpu=5,
_temp_dir='/scratch/leuven/330/vsc33053/ray_spill',
split_pattern = '-')
2024-03-06 17:17:33,892 INFO worker.py:1724 -- Started a local Ray instance.
2024-03-06 17:17:34,368 cisTopic INFO Subsetting data for AST (30 of 2853)
2024-03-06 17:17:36,523 cisTopic INFO Computing p-value for AST
2024-03-06 17:17:40,292 cisTopic INFO Computing log2FC for AST
2024-03-06 17:17:41,563 cisTopic INFO AST done!
2024-03-06 17:17:41,564 cisTopic INFO Subsetting data for BG (283 of 2853)
2024-03-06 17:17:41,588 cisTopic INFO Computing p-value for BG
2024-03-06 17:17:44,487 cisTopic INFO Computing log2FC for BG
2024-03-06 17:17:44,500 cisTopic INFO BG done!
2024-03-06 17:17:44,502 cisTopic INFO Subsetting data for COP (66 of 2853)
2024-03-06 17:17:44,527 cisTopic INFO Computing p-value for COP
2024-03-06 17:17:47,519 cisTopic INFO Computing log2FC for COP
2024-03-06 17:17:47,533 cisTopic INFO COP done!
2024-03-06 17:17:47,535 cisTopic INFO Subsetting data for ENDO (16 of 2853)
2024-03-06 17:17:47,566 cisTopic INFO Computing p-value for ENDO
2024-03-06 17:17:50,431 cisTopic INFO Computing log2FC for ENDO
2024-03-06 17:17:50,469 cisTopic INFO ENDO done!
2024-03-06 17:17:50,471 cisTopic INFO Subsetting data for GC (45 of 2853)
2024-03-06 17:17:50,486 cisTopic INFO Computing p-value for GC
2024-03-06 17:17:53,347 cisTopic INFO Computing log2FC for GC
2024-03-06 17:17:53,366 cisTopic INFO GC done!
2024-03-06 17:17:53,368 cisTopic INFO Subsetting data for GP (16 of 2853)
2024-03-06 17:17:53,381 cisTopic INFO Computing p-value for GP
2024-03-06 17:17:56,200 cisTopic INFO Computing log2FC for GP
2024-03-06 17:17:56,239 cisTopic INFO GP done!
2024-03-06 17:17:56,241 cisTopic INFO Subsetting data for INH_PVALB (15 of 2853)
2024-03-06 17:17:56,255 cisTopic INFO Computing p-value for INH_PVALB
2024-03-06 17:17:59,070 cisTopic INFO Computing log2FC for INH_PVALB
2024-03-06 17:17:59,107 cisTopic INFO INH_PVALB done!
2024-03-06 17:17:59,108 cisTopic INFO Subsetting data for INH_SNCG (38 of 2853)
2024-03-06 17:17:59,251 cisTopic INFO Computing p-value for INH_SNCG
2024-03-06 17:18:02,159 cisTopic INFO Computing log2FC for INH_SNCG
2024-03-06 17:18:02,209 cisTopic INFO INH_SNCG done!
2024-03-06 17:18:02,210 cisTopic INFO Subsetting data for INH_SST (58 of 2853)
2024-03-06 17:18:02,226 cisTopic INFO Computing p-value for INH_SST
2024-03-06 17:18:05,034 cisTopic INFO Computing log2FC for INH_SST
2024-03-06 17:18:05,048 cisTopic INFO INH_SST done!
2024-03-06 17:18:05,050 cisTopic INFO Subsetting data for INH_VIP (77 of 2853)
2024-03-06 17:18:05,069 cisTopic INFO Computing p-value for INH_VIP
2024-03-06 17:18:07,901 cisTopic INFO Computing log2FC for INH_VIP
2024-03-06 17:18:07,955 cisTopic INFO INH_VIP done!
2024-03-06 17:18:07,957 cisTopic INFO Subsetting data for MG (63 of 2853)
2024-03-06 17:18:07,972 cisTopic INFO Computing p-value for MG
2024-03-06 17:18:10,843 cisTopic INFO Computing log2FC for MG
2024-03-06 17:18:10,860 cisTopic INFO MG done!
2024-03-06 17:18:10,861 cisTopic INFO Subsetting data for MGL (65 of 2853)
2024-03-06 17:18:10,876 cisTopic INFO Computing p-value for MGL
2024-03-06 17:18:13,720 cisTopic INFO Computing log2FC for MGL
2024-03-06 17:18:13,735 cisTopic INFO MGL done!
2024-03-06 17:18:13,736 cisTopic INFO Subsetting data for MOL (745 of 2853)
2024-03-06 17:18:13,758 cisTopic INFO Computing p-value for MOL
2024-03-06 17:18:16,621 cisTopic INFO Computing log2FC for MOL
2024-03-06 17:18:16,634 cisTopic INFO MOL done!
2024-03-06 17:18:16,636 cisTopic INFO Subsetting data for NFOL (279 of 2853)
2024-03-06 17:18:16,654 cisTopic INFO Computing p-value for NFOL
2024-03-06 17:18:19,518 cisTopic INFO Computing log2FC for NFOL
2024-03-06 17:18:19,535 cisTopic INFO NFOL done!
2024-03-06 17:18:19,538 cisTopic INFO Subsetting data for OPC (170 of 2853)
2024-03-06 17:18:19,556 cisTopic INFO Computing p-value for OPC
2024-03-06 17:18:22,411 cisTopic INFO Computing log2FC for OPC
2024-03-06 17:18:22,439 cisTopic INFO OPC done!
2024-03-06 17:18:22,441 cisTopic INFO Subsetting data for PURK (10 of 2853)
2024-03-06 17:18:22,457 cisTopic INFO Computing p-value for PURK
2024-03-06 17:18:25,299 cisTopic INFO Computing log2FC for PURK
2024-03-06 17:18:25,317 cisTopic INFO PURK done!
[171]:
plot_imputed_features(
cistopic_obj,
reduction_name='UMAP',
imputed_data=gene_act,
features=['PDGFRA', 'OLIG2', 'MBP', 'SOX10', # Olig differentiation
'CTNNA3', 'ENPP6', 'OLIG1', # Olig differentiation
'GAD2', 'VIP', 'SST', 'CTXN3', # Int
'NFIB', 'SOX9', #Ast
'LEF1', #Endo
'SPI1'], #Glia
scale=True,
num_columns=4
)
[172]:
print("Number of DAGs found:")
print("---------------------")
for x in markers_dict:
print(f" {x}: {len(DAG_markers_dict[x])}")
Number of DAGs found:
---------------------
AST: 2321
BG: 2668
COP: 586
ENDO: 2583
GC: 2816
GP: 2948
INH_PVALB: 2683
INH_SNCG: 2736
INH_SST: 2786
INH_VIP: 2714
MG: 2256
MGL: 4022
MOL: 1735
NFOL: 704
OPC: 2487
PURK: 2650
Label transfer
Exploting the gene activity scores, we can transfer labels from a reference data set (e.g. scRNA-seq). As an example, we will transfer labels from the scRNA-seq layer of this data set.
The methods available for label transferring are: - ingest (from scanpy) - harmony (Korsunsky et al, 2019) - bbknn (Polański et al, 2020) - scanorama (Hie et al, 2019) - cca.
Except for ingest, these methods return a common coembedding and labels are inferred using the distances between query and refenrence cells as weights.
[185]:
from pycisTopic.label_transfer import label_transfer
[199]:
import scanpy as sc
rna_anndata = sc.read_h5ad(
"/staging/leuven/stg_00002/lcb/sdewin/PhD/python_modules/scenicplus_development_tutorial/scRNA_seq_pp/adata.h5ad"
).raw.to_adata()
atac_anndata = sc.AnnData(gene_act.mtx.T, obs = pd.DataFrame(index = gene_act.cell_names), var = pd.DataFrame(index = gene_act.feature_names))
[200]:
atac_anndata.obs["sample_id"] = "10x_multiome_brain"
rna_anndata.obs["sample_id"] = "10x_multiome_brain"
[204]:
label_dict = label_transfer(
rna_anndata,
atac_anndata,
labels_to_transfer = ['Seurat_cell_type'],
variable_genes = True,
methods = ['ingest', 'harmony', 'bbknn', 'scanorama', 'cca'],
return_label_weights = False,
_temp_dir= '/scratch/leuven/330/vsc33053/ray_spill/'
)
2024-03-06 17:30:35,512 cisTopic INFO Normalizing RNA data
WARNING: adata.X seems to be already log-transformed.
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/scanpy/preprocessing/_simple.py:843: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
2024-03-06 17:30:41,665 cisTopic INFO Processing 1 query sample(s) using 1 cpu(s)
2024-03-06 17:30:44,886 INFO worker.py:1724 -- Started a local Ray instance.
(label_transfer_ray pid=3796267) 2024-03-06 17:30:47,417 cisTopic INFO Normalizing ATAC data from sample 10x_multiome_brain
(label_transfer_ray pid=3796267) /lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/scanpy/preprocessing/_simple.py:843: UserWarning: Received a view of an AnnData. Making a copy.
(label_transfer_ray pid=3796267) view_to_actual(adata)
(label_transfer_ray pid=3796267) /lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/pycisTopic/label_transfer.py:237: FutureWarning: Use anndata.concat instead of AnnData.concatenate, AnnData.concatenate is deprecated and will be removed in the future. See the tutorial for concat at: https://anndata.readthedocs.io/en/latest/concatenation.html
(label_transfer_ray pid=3796267) adata_concat = ref_anndata.concatenate(
(label_transfer_ray pid=3796267) 2024-03-06 17:30:55,619 cisTopic INFO Running integration with ingest
(label_transfer_ray pid=3796267) /lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/umap/umap_.py:1943: UserWarning: n_jobs value -1 overridden to 1 by setting random_state. Use no seed for parallelism.
(label_transfer_ray pid=3796267) warn(f"n_jobs value {self.n_jobs} overridden to 1 by setting random_state. Use no seed for parallelism.")
(label_transfer_ray pid=3796267) <frozen _collections_abc>:949: ImplicitModificationWarning: Setting element `.obsm['rep']` of view, initializing view as actual.
(label_transfer_ray pid=3796267) <frozen _collections_abc>:949: ImplicitModificationWarning: Setting element `.obsm['X_umap']` of view, initializing view as actual.
(label_transfer_ray pid=3796267) <frozen _collections_abc>:949: ImplicitModificationWarning: Setting element `.obsm['X_pca']` of view, initializing view as actual.
(label_transfer_ray pid=3796267) /lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/pycisTopic/label_transfer.py:247: FutureWarning: In a future version, `df.iloc[:, i] = newvals` will attempt to set the values inplace instead of always setting a new array. To retain the old behavior, use either `df[df.columns[i]] = newvals` or, if columns are non-unique, `df.isetitem(i, newvals)`
(label_transfer_ray pid=3796267) query_anndata.obs.loc[:, var] = query_anndata.obs.loc[
(label_transfer_ray pid=3796267) 2024-03-06 17:31:28,883 cisTopic INFO Running integration with harmony
(label_transfer_ray pid=3796267) 2024-03-06 17:31:29,391 harmonypy INFO Computing initial centroids with sklearn.KMeans...
(label_transfer_ray pid=3796267) 2024-03-06 17:31:29,391 - harmonypy - INFO - Computing initial centroids with sklearn.KMeans...
(label_transfer_ray pid=3796267) 2024-03-06 17:31:30,685 harmonypy INFO sklearn.KMeans initialization complete.
(label_transfer_ray pid=3796267) 2024-03-06 17:31:30,697 harmonypy INFO Iteration 1 of 10
(label_transfer_ray pid=3796267) 2024-03-06 17:31:30,685 - harmonypy - INFO - sklearn.KMeans initialization complete.
(label_transfer_ray pid=3796267) 2024-03-06 17:31:30,697 - harmonypy - INFO - Iteration 1 of 10
(label_transfer_ray pid=3796267) 2024-03-06 17:31:31,458 harmonypy INFO Iteration 2 of 10
(label_transfer_ray pid=3796267) 2024-03-06 17:31:31,458 - harmonypy - INFO - Iteration 2 of 10
(label_transfer_ray pid=3796267) 2024-03-06 17:31:32,189 harmonypy INFO Iteration 3 of 10
(label_transfer_ray pid=3796267) 2024-03-06 17:31:32,189 - harmonypy - INFO - Iteration 3 of 10
(label_transfer_ray pid=3796267) 2024-03-06 17:31:32,916 harmonypy INFO Iteration 4 of 10
(label_transfer_ray pid=3796267) 2024-03-06 17:31:32,916 - harmonypy - INFO - Iteration 4 of 10
(label_transfer_ray pid=3796267) 2024-03-06 17:31:33,682 harmonypy INFO Converged after 4 iterations
(label_transfer_ray pid=3796267) 2024-03-06 17:31:33,722 cisTopic INFO Running integration with bbknn
(label_transfer_ray pid=3796267) 2024-03-06 17:31:33,682 - harmonypy - INFO - Converged after 4 iterations
(label_transfer_ray pid=3796267) WARNING: consider updating your call to make use of `computation`
(label_transfer_ray pid=3796267) 2024-03-06 17:31:45,983 cisTopic INFO Running integration with scanorama
(label_transfer_ray pid=3796267) Found 1430 genes among all datasets
(label_transfer_ray pid=3796267) [[0. 0.34805468]
(label_transfer_ray pid=3796267) [0. 0. ]]
(label_transfer_ray pid=3796267) Processing datasets (0, 1)
(label_transfer_ray pid=3796267) 2024-03-06 17:31:50,070 cisTopic INFO Running integration with cca
We can now add the annotations to our cisTopic object and visualize them in the cell-topic UMAP. Scanorama and harmony are the ones that work best.
[205]:
label_dict_x=[label_dict[key] for key in label_dict.keys()]
label_pd = pd.concat(label_dict_x, axis=1, sort=False)
label_pd.index = cistopic_obj.cell_names
label_pd.columns = ['pycisTopic_' + x for x in label_pd.columns]
cistopic_obj.add_cell_data(label_pd, split_pattern = '-')
[207]:
plot_metadata(
cistopic_obj,
reduction_name='UMAP',
variables=['Seurat_cell_type'] + label_pd.columns.to_list(),
remove_nan=True,
seed=555,
num_columns=3)
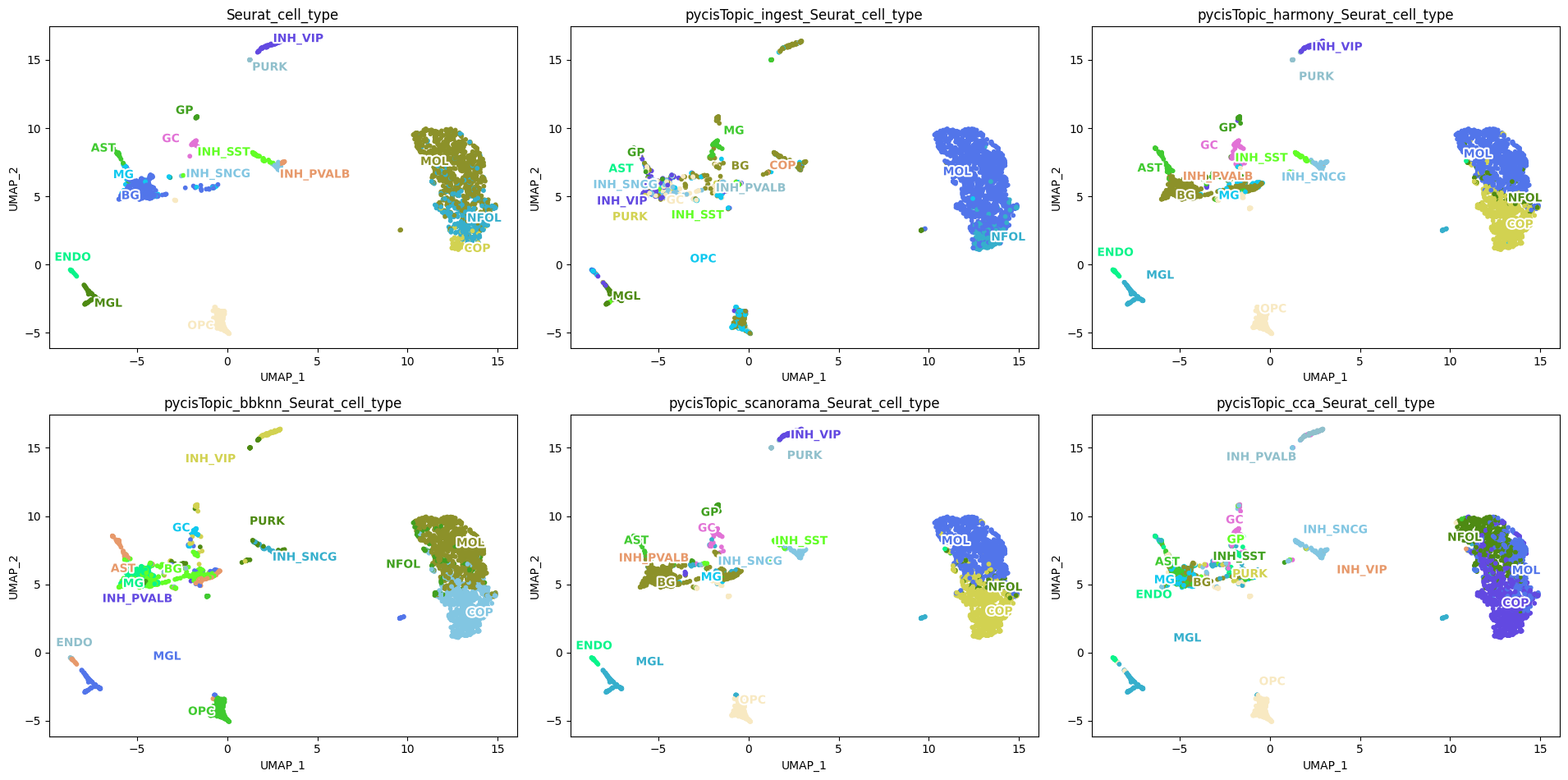
Let’s plot a confusion matrix.
[236]:
import seaborn as sns
fig, axs = plt.subplots(ncols = 3, nrows = 2, figsize = (3 * 5, 2 * 5))
for method, ax in zip(label_pd.columns.to_list(), axs.ravel()):
conf_mat = pd.crosstab(cistopic_obj.cell_data["Seurat_cell_type"], cistopic_obj.cell_data[method])
conf_mat = conf_mat / conf_mat.sum()
sns.heatmap(conf_mat.loc[conf_mat.columns], ax = ax)
fig.tight_layout()
fig.show()
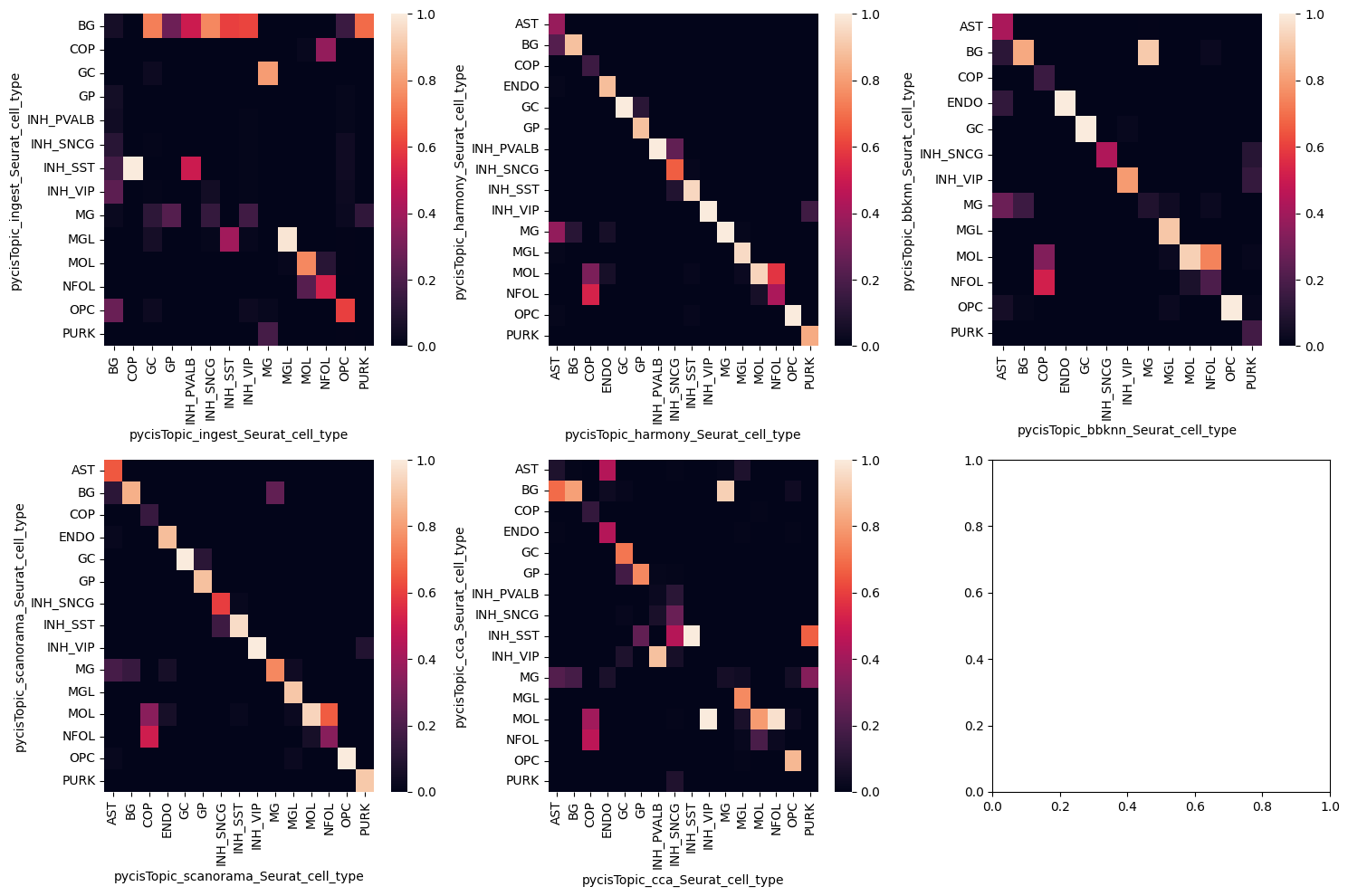
Exporting to loom
We can now create loom files to further explore the results. There are two types of loom files:
Region accessibility: These loom files include the imputed accessibility as matrix, topics as regulons and cell-topic distributions as AUC matrices. The imputed values, the cistopic object used for the imputation and the cell-topic and topic-region binarized distributions are required. Alternatively, we can also provide different clustering and the marker regions (DARs) per group per clustering.
Gene activity: These loom files contain the gene activity as matrix, scRNA-seq derived regulons and their AUC values based on gene activity. The gene activity values, the cistopic object, and scRNA-seq derived regulons are required. Alternatively, we can also provide different clustering and the marker genes (DAGs) per group per clustering.
[240]:
from pycisTopic.loom import export_region_accessibility_to_loom, export_gene_activity_to_loom
[241]:
cluster_markers = {'Seurat_cell_type': markers_dict}
[242]:
os.makedirs(os.path.join(out_dir, "loom"), exist_ok=True)
[244]:
export_region_accessibility_to_loom(
accessibility_matrix = imputed_acc_obj,
cistopic_obj = cistopic_obj,
binarized_topic_region = region_bin_topics_otsu,
binarized_cell_topic = binarized_cell_topic,
selected_cells = cistopic_obj.projections['cell']['UMAP'].index.tolist(),
out_fname = os.path.join(out_dir, "loom", "10x_multiome_brain_pycisTopic_region_accessibility.loom"),
cluster_annotation = ['Seurat_cell_type'],
cluster_markers = cluster_markers,
tree_structure = ('10x_multiome_brain', 'pycisTopic', 'noDBL_all'),
title = 'Tutorial - Region accessibility all',
nomenclature = "hg38",
split_pattern = '-'
)
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/pycisTopic/loom.py:619: FutureWarning: In a future version, `df.iloc[:, i] = newvals` will attempt to set the values inplace instead of always setting a new array. To retain the old behavior, use either `df[df.columns[i]] = newvals` or, if columns are non-unique, `df.isetitem(i, newvals)`
regulon_mat.loc[:, col_idx] = np.where(
2024-03-06 18:10:04,308 cisTopic INFO Creating minimal loom
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/pycisTopic/loom.py:840: FutureWarning: iteritems is deprecated and will be removed in a future version. Use .items instead.
for name, threshold in auc_thresholds.iteritems()
2024-03-06 18:11:27,242 cisTopic INFO Adding annotations
2024-03-06 18:11:49,942 cisTopic INFO Adding clusterings
2024-03-06 18:11:49,980 cisTopic INFO Adding markers
No markers for nan
2024-03-06 18:12:01,188 cisTopic INFO Exporting
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/loomxpy/loomxpy.py:459: FutureWarning: The default value of regex will change from True to False in a future version.
regulons.columns = regulons.columns.str.replace("_?\\(", "_(")
/lustre1/project/stg_00002/mambaforge/vsc33053/envs/pycisTopic_polars_tutorial/lib/python3.11/site-packages/loomxpy/loomxpy.py:437: FutureWarning: The default value of regex will change from True to False in a future version.
regulons.columns = regulons.columns.str.replace("_?\\(", "_(")
[246]:
export_gene_activity_to_loom(
gene_activity_matrix = gene_act,
cistopic_obj = cistopic_obj,
out_fname = os.path.join(out_dir, "loom", "10x_multiome_brain_pycisTopic_gene_activity.loom"),
cluster_annotation = ['Seurat_cell_type'],
cluster_markers = cluster_markers,
tree_structure = ('10x_multiome_brain', 'pycisTopic', 'ATAC'),
title = 'Tutorial - Gene activity',
nomenclature = "hg38",
split_pattern = '-'
)
2024-03-06 18:13:30,346 cisTopic INFO Creating minimal loom
2024-03-06 18:13:39,158 cisTopic INFO Adding annotations
2024-03-06 18:13:41,059 cisTopic INFO Adding clusterings
2024-03-06 18:13:41,095 cisTopic INFO Adding markers
No markers for nan
2024-03-06 18:13:41,482 cisTopic INFO Exporting